Received Date:
24 December 2019 Revised Date:
29 September 2020 Available Online:
10 November 2020
Abstract:
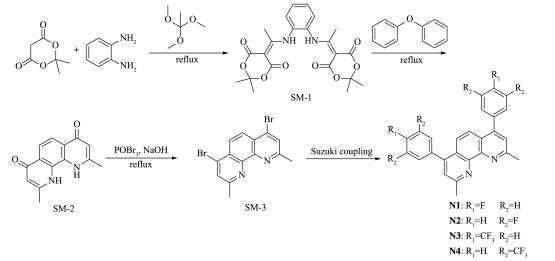
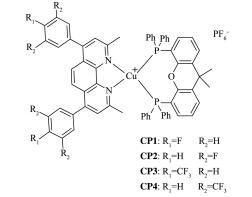
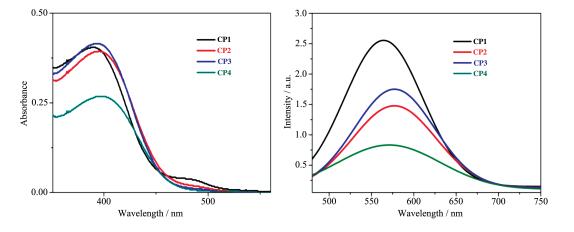
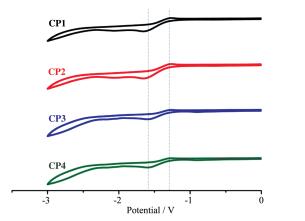
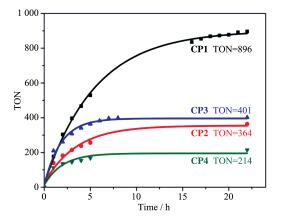
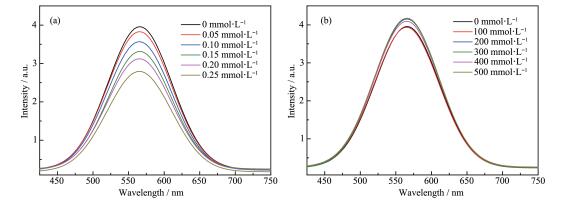
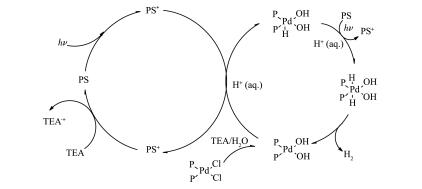
A series of novel bidentate ligands of fluoro phenanthrolines were designed and synthesized, which could formulate a series of heteroleptic copper photosensitizers CP1~CP4 with Cu(MeCN)4PF6 and Xantphos as P ligand. The photosensitive activities of this copper complex were researched in water reduction system, and the turnover number (TON) of hydrogen evolution was up to 896. The absorption spectrum and fluorescence emission spectrum of the copper complexes indicated the good stability in solution. The oxidation quenching is the main quenching pathway in water reduction system, which was confirmed by the fluorescence quenching experiments. Moreover, a preliminary explanation and discussion of the structure-activity relationship and the mechanism of photocatalytic hydrogen evolution from water were carried out.
(a) Hisatomi T, Kubota J, Domen K. Chem. Soc. Rev., 2014, 43: 7520-7535 (b)Berardi S, Drouet S, Llobet A, et al. Chem. Soc. Rev., 2014, 43: 7501-7519
[2]
Esswein A J, Nocera D G. Chem. Rev., 2007, 107(10):4022-4047 doi: 10.1021/cr050193e
[3]
(a) Kalyanasundaram K, Kiwi J, Grätzel M. Helv. Chim. Acta, 1978, 61: 2720-2730 (b)Kirch M, Lehn J M, Sauvage J P. Helv. Chim. Acta, 1979, 62: 1345-1384 (c)Kiwi J, Gratzel M. J. Am. Chem. Soc., 1978, 100(20): 6314-6320
[4]
(a) Abbotto A, Manfredi N. Dalton Trans., 2011, 40: 12421-12438 (b)Ganga G L, Puntoriero F, Campagna S, et al. Faraday Discuss., 2012, 155: 177-190 (c)Deponti E, Natali M. Dalton Trans., 2016, 45: 9136-914 (d)Lin H, Liu D, Wang X X, et al. Phys. Chem. Chem. Phys., 2015, 17: 10726-10736 (e)Na Y, Wei P C, Zhou L. Chem. Eur. J., 2016, 22: 10365-10368
[5]
(a) Jiang W N, Liu J H, Li C. Inorg. Chem. Commun., 2012, 16: 81-85 (b)Zhou R W, Manbeck G F, Brewer K J, et al. Chem. Commun., 2015, 51: 12966-12969 (c)Mengele A K, Kaufhold S, Rau S, et al. Dalton Trans., 2016, 45: 6612-6618
[6]
(a) Du P W, Knowles K, Eisenberg R. J. Am. Chem. Soc., 2008, 130(38): 12576-12577 (b)Wang C J, Chen Y, Fu W F. Dalton Trans., 2015, 44: 14483-14493 (c)Whang D R, Park S Y. ChemSusChem, 2015, 8: 3204-3207 (d)Kitamoto K, Sakai K. Chem. Commun., 2016, 52: 1385-1388
[7]
(a) Disalle B F, Bernhard S. J. Am. Chem. Soc., 2011, 133(31): 11819-11821 (b)Gärtner F, Denurra S, Beller M, et al. Chem. Eur. J., 2012, 18: 3220-3225 (c)Lu Y, McGoldrick N, Murphy F, et al. Chem. Eur. J., 2016, 22(32): 11349-11356 (d)Xu D N, Chu Q Q, Fang B Z, et al. J. Catal., 2015, 325: 118-127
[8]
Zhang X J, Jin Z L, Li Y X, et al. J. Phys. Chem. C, 2009, 113(6):2630-2635 doi: 10.1021/jp8085717
[9]
(a)Probst B, Guttentag M, Rodenberg A, et al. Inorg. Chem., 2011,50(8):3404-3412 (b)Du P, Schneider J, Li F, et al. J. Am. Chem. Soc., 2008, 130(15):5056-5058
[10]
Horiuchi Y, Toyao T, Saito M, et al. J. Phys. Chem. C, 2012, 116(39):20848-20853 doi: 10.1021/jp3046005
(a) Zhang W, Hong J D, Zheng J W, et al. J. Am. Chem. Soc.,2011, 133(51): 20680-20683 (b)Lazarides T, Mccormick T, Du P, et al. J. Am. Chem. Soc., 2009, 131(26): 9192-9194 (c)Mccormick T M, Calitree B, Orchard A, et al. J. Am. Chem. Soc., 2010, 132(44): 15480-15483 (d)Chan S F, Chou M, Creutz C, et al. J. Am. Chem. Soc., 1981, 103(2): 369-379
[13]
(a) Huang G L, Shi R, Zhu Y F. J. Mol. Catal. A: Chem., 2011, 348: 100-105 (b)He X D, Yin L X, Li Y Q. New J. Chem., 2019, 43: 6577-6586 (c)Gu L Y, Lei Y, Luo J, et al. ACS Appl. Mater. Interfaces., 2019, 11: 24789-24794 (d)Yang H M, Guo M M, Hu X Y, et al. Appl. Surf. Sci., 2019, 494: 501-507
[14]
(a) Chen N Y, Xia L M, Sun Y Y, et al. Chem. Eur. J., 2017, 23(15): 3631-3636
(b)WU Qing-An(吴庆安), CHEN Hao(陈浩), HUANG Dao-Chen(黄道臣), et al. Chinese J. Inorg. Chem.(无机化学学报), 2018, 34: 1-7
(c)XIA Liang-Min(夏良敏), CHEN Hao(陈浩), WU Qing-An (吴庆安), et al. Chinese J. Inorg. Chem.(无机化学学报), 2017, 33(4): 560-568
[15]
Larsen A F, Ulven T. Org. Lett., 2011, 13(13):3546-3548 doi: 10.1021/ol201321z
[16]
Zhao Y F, Schwab M G, Kiersnowski A, et al. J. Mater. Chem. C, 2016, 4:4640-4646 doi: 10.1039/C6TC00780E
[17]
(a) Luo S P, Mejía E, Friedrich A, et al. Angew. Chem. Int. Ed., 2013, 52(1): 419-423 (b)Luo S P, Chen N Y, Sun Y Y, et al. Dyes Pigm., 2016, 134: 580-585
[18]
Knorn M, Rawner T, Czerwieniec R, et al. ACS Catal., 2015, 5(9):5186-5193 doi: 10.1021/acscatal.5b01071

 下载:
下载:

 下载:
下载:








