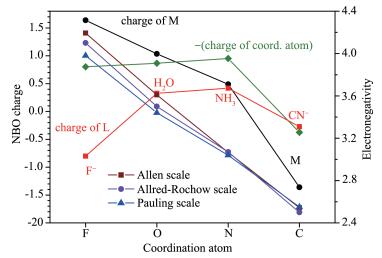
Figure 1.
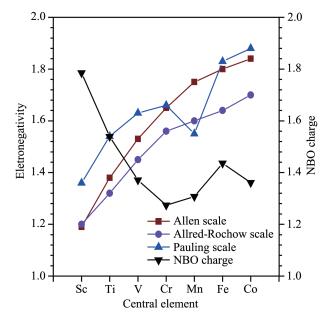
Trend of NBO charge on central cobalt atom, -(charge of coordinated atoms), and each ligand compared with three scale (Pauling's, Allred-Rochow's and Allen's) electronegativity of coordination atoms
Verifying Principle of Electroneutrality by NBO Charge Calculations
Jia-Wei XU , Li-Ke DENG , Hao-Zhe ZHANG , Huan-Yu ZHANG , Yu-Chen LUO , Xin-Ya YAN , Jian-Chun Bao , Min FANG , Yong WU

It is well - known that free ions, such as Cu2+, is unstable in aqueous solution, having a strong tendency to form hydrate ions[1-2]. In non - aqueous solvent, they will also coordinate with solvent molecules. This phe-nomenon is the result of electrostatic properties of mol-ecules or ions, which respond to the environment through polarization by the local electric field[3]. Ions with smaller radii and higher charges have strong polar-izing ability, while ions with larger radii will be easier to be polarized. Hard-soft acid and base (HSAB) theory rooted in these concepts[4-7].
To evaluate polarization capacities of ions[8-9], Cart-ledge coined the concept of ionic potential (φ=Z/r) where Z is the charge of an ion and r represents its radius. Later, Xu proposed to use Z2/r to evaluate the polarization capacities of ions[10-13]. These parameters have advantages in quantitative calculations and explained experimental results commendably, having been accepted widely in high-level chemistry educa-tion and scientific studies nowadays. One consequence of polarization is that electrons are redistributed to reduce the electrostatic force between two entities when approaching to form a chemical bond. Thus, the polarization effect will reduce the charge of the ions and contribute to the covalent bonding of the bond. This is the reason why absolute ionic or covalent bonds between two different elements do not exist[14-16]. Only in some nonpolar covalent ones, bonding electrons are equally distributed between two atoms, and pure cova-lent bonds are formed. A chemical bond usually has some ionic character and some covalent character. The strength of a chemical bond is the results of both contri-butions.
Early in 1948, Pauling proposed the principle of electroneutrality (EN)."It has seemed to me likely that in general all of the atoms in the complexes that consti-tute stable chemical substances have resultant electri-cal charges smaller than those shown by these most electropositive and electronegative atoms in their com-pounds with one another, and I have accordingly formu-lated the postulate of the essential electrical neutrality of atoms: namely, that the electronic structure of sub-stances is such as to cause each atom to have essentially zero resultant electrical charge, the amount of leeway being not greater than about ±1/2, and these resultant charges are possessed mainly by the most electroposi-tive and electronegative atoms, and are distributed in such a way as to correspond to electrostatic ability"[17]. Huheey et al. wrote in the Inorganic Chemistry text-book, "Pauling suggested that complexes would be most stable when the electronegativity of the ligand was such that the metal achieved a condition of essen-tially zero net electrical charge. This tendency for zero or low electrical charges on atoms is a rule -of -thumb known as the electroneutrality principle, and it is used to make predictions regarding electronic structure in many types of compounds, not only complexes "[18]. Crabtree in his Organometallic Chemistry text book wrote about the EN principle, "Linus Pauling (1901— 1994), a giant of twentieth century chemistry, proposed the electroneutrality principle in which electrons dis-tribute themselves in polar covalent molecules so that each atomic charge is nearly neutral. In practice, these charges fall in a range from about +1 to -1. The non-metals N, O, and F tend to be negatively charged while metals such as Na or Al are positively charged"[19]. We take Crabtree's definition of the EN principle as the advanced version of the originally proposed version. This definition should have been widely accepted since the textbook written by Crabtree is well circulated[19-21]. This principle was said to be very powerful [21], and has been used to explain experimental results[18]. GarcíaLastra et al. in their research paper stated:"we propose that the color change in the Al2O3·xCr2O3 series is mainly related to the EN principle by Pauling for a transition-metal complex, stating that the total charge of the transition - metal cation in the complex is nearly zero"[22].
Contradictions were also reported and written in textbook, "In apparent contradiction to the electroneu-trality principle, there are many complexes in which the metal exists in a low oxidation state and yet is bond-ed to an element of fairly low electronegativity. Among the most prominent examples are the transition metal carbonyls, a large class of compounds in which the ligand (CO) is bound to the central metal through car-bon. The source of stability in these complexes is the capacity of the carbon monoxide ligand to accept a back donation of electron density from the metal atom"[18]. Although the EN principle applies for bonds in compounds formed by elements with low oxidation numbers, whether this principle is also applicable when it comes to compounds containing elements with high formal oxidation numbers, and what are the affect-ing factors? These questions have not been answered.
Development in computational chemistry makes it possible for us to study charge distribution in mole-cules. Using natural - population - analysis to obtain the NBO charge, also called the natural atomic charge, to measure the net charge of a specific atom in a molecule or an ion has been widely applied[23-31] and show superi-ority compared to other methods[59-61]. Density function-al theory (DFT) of electronic structure has made an un-paralleled impact on the application of quantum me-chanics to interesting and challenging problems in chemistry[32]. B3LYP is the most popular and the most widely used of all the DFT functions. It has enjoyed a remarkable performance over a wide range of sys-tems[32]. Batista et al. found that by sufficiently expand-ing the basis set of the ligands coordinated to the metal ions with polarization functions, the hybrid B3LYP function predicts equilibrium distances and exchange coupling constants of pre-selected spin- electronic states are in excellent agreement with X-ray and mag-netic data[33].
Hence, in this work, NBO charges of hexahydrate trivalent ions of [M(H2O)6]3+ (M=Sc, Ti, V, Cr, Mn, Fe, Co) and [Co(Ⅲ) L6] (L=F-, H2O, NH3, CN-) were calculated at B3LYP/6-31++G(d, p) level of DFT. In order to simulate the charges of Al and O atoms in α-Al2O 3, two Al cluster compounds, Al3O(OH)7(H2O)5 (Al3) and Al6O6(OH)6(H2O)5 (Al6) were created based on the crystal structure of α-Al2O3[34] and their NBO charges were cal-culated at B3LYP/6 - 31++G(d, p) level of theory. The results are that except for [Co(NH3)6 ]3+, they all violate the EN principle by having charges of atoms out of the range of +1 to -1. Contrary to the EN principle, we find that the charge of an element is not determined by the element alone, but can be enormously influenced by the type of its immediately adjacent bonding atoms.
Here, all calculations for transition metal complexes are carried out with not only polarization functions, but also dispersion functions using Gaussian 09 program[35] at B3LYP/6-31++G(d, p) level of DFT on preselected spin-multiplicity of metal ions based on experimental evidence or arguments. All electrons of metal ions were considered without using effective core potentials (ECPs) and calculated using 6 -31++G(d, p) basis set. All calculated structures are true minima, i.e., no imaginary frequency was observed.
Parts of the crystal structure of α-Al2O3 were selected and the dangling bonds were terminated with OH- or H2O, resulting in neutral Al3O(OH)7(H2O)5 (Al3) and Al6O6(OH)6(H2O)5 (Al6). The structures of these two compounds were optimized at B3LYP/6-31G(d, p) level of DFT and found to be the true minima. The NBO charges of atoms of the optimized structures were calcu-lated at the same level of theory.
2.1 [Co(Ⅲ) L6]3- (L=F-, H2O, NH3, CN-) and the accuracy of the calculation method
Effects of different coordination atoms on the NBO charges of the central metal ion were studied by calculating four [Co(Ⅲ) L6 ]3- complexes (only [CoF6]3-were calculated as a high-spin cation, others as low-spin cations) and the results are given in Fig. 1 and Table 1.
 下载:
导出CSV
下载:
导出CSV
| NBO charge | Electronegativity of coordination atoms | Bond length (M-O) / nm | |||||
| L | Co | -(coord. atom)* | L | Allen | Pauling | Allred-Rochow | |
| F- | 1.639 | 0.802 | -0.802 | 4.19 | 3.98 | 4.10 | 0.206 |
| H2O | 1.034 | 0.865 | 0.327 | 3.61 | 3.44 | 3.50 | 0.192 |
| NH3 | 0.489 | 0.952 | 0.418 | 3.07 | 3.04 | 3.07 | 0.203 |
| CN- | -1.360 | -0.375 | -0.273 | 2.54 | 2.55 | 2.50 | 0.195 |
| *Charge divided by -1. | |||||||
The optimized geometries of [CoF6]3- and [Co(CN)6]3- are given in Fig. 2. The calculated Co-C bond length was 0.195 nm for [Co(CN)6]3-, consistent with the experimental value in the solid state (0.191 nm)[36]. It is also known that embedding the complex in a condensed phase lowers the orbital energies and sta-bilizes the system and the M - L bond lengths would be expected to shorten[37]. Since all calculations treat the complexes as gas phase molecules, the bond distance lengthening is consistent with the above observation.
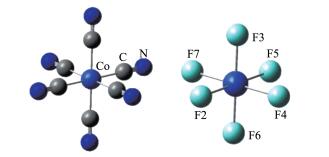
Co-C: 0.195 0 nm; Co-F2, F4, F5 or F7: 0.205 5 nm; Co-F3 or F6:0.191 4 nm
Cotton and Meyers[38] have measured the magnetic moments of K3[CoF6] and Ba3[CoF 6]2 to be 5.25 B.M.~ 5.66 B.M. in a temperature range of 73~299 K, mean-ing [CoF6]3- are high spin and have four unpaired elec-trons. The electronic spectra of various [CoF6]3- salts suggest that they have static or dynamic Jahn- Teller effect in its structure[38-39]. The calculated Co-F bond lengths were 0.191 4 nm (two bonds along z-axis) and 0.205 5 nm, which are in excellent agreement with the distances (0.194 5 nm (two bonds along z-axis) and 0.206 5 nm) of the optimized geometry at B3LYP meth-od with def2 - TZVP basis set and Stuttgart RSC 1997 Effective Core Potential by Monajjemi and Khaleghi-an[40]. They also detected the Jahn - Teller distortion of the high-spin d6 electronic configuration.
The only available experimental bond distance of Co-F in [CoF6]3- is obtained as 0.189 nm in CoF3 struc-tures[41], where Co is six coordinated and thus CoF3 can be viewed as [CoF6]3-. The structural determination is based on the powder X-ray diffraction pattern deter-mined by Peacock et al. in 1957 assuming it has an octahedral geometry. Their work and a paper by Babel[42] were sometimes wrongly cited as providing structural information for Co-F distance in the K3[CoF6] crystal. Thus, the experimental bond distances of Co-F bond distances in [CoF6]3- still need to be determined, and better determined based on single-crystal XRD dif-fraction data.
[Co(H2O)6]3+ is found to have a low - spin ground state[43-49]. Winkler et al. estimated the energy differ-ence between the low and high spin state is 17.6~19.2 kJ·mol-1 [48]. Navon estimated that the lower limit for the difference in the free energies of the low -spin and high - spin states of [Co(H2O)6]3+ is ΔG⊖ > 22.6 kJ·mol-1 based on 59Co NMR studies and pointed out that high-spin state are thermally accessible[49]. ΔG⊖ were calculated to be (38.1±15.9) kJ·mol-1 by Johnson and Nelson based on thermodynamic data[50]. Consistent with the reported results, our calculations also show that high-spin and low spin [Co(H2O)6]3+ have very similar energy. However, our calculations wrongly predicted that high-spin was the ground state (free energy: -1 840.046 375 Hartree), 14.6 kJ·mol-1 more stable than the low -spin ground state (-1 840.040 840 Har-tree). Batista et al.[33] reported that the energetics of low-lying spin-states is beyond the capabilities of the DFT/B3LYP level. However, they found that the hybrid B3LYP function predicts equilibrium distances and exchange coupling constants of pre -selected spin-electronic states in excellent agreement with X-ray and magnetic data. The above - mentioned limitation of the DFT/B3LYP level method does not prevent it from being successfully applied in NBO charge calcula-tions[29-31].
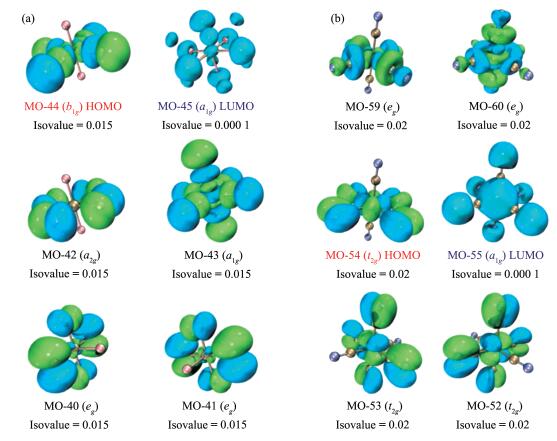
It can be seen that from the most positive Co charge in [CoF6]3- (1.639) to the most negative Co in [Co(CN)6]3- (-1.360), a large difference (3.0) of charge appears (Fig. 1). Consistently, the diagrams of frontier occupied MOs of [CoF6]3- (Fig. 3a) shows little or no electron density at Co, but a lot in the diagrams of fron-tier occupied MOs of [CoF6]3- (Fig. 3b). Only Co (0.489) in [Co(NH3)6]3+ falls in a range of -1 to +1. Thus, the charge distributions in [CoF6]3-, [Co(H2O)6]3+ and [Co(CN)6]3- violate the EN principle.
By checking the electron density, ionic or cova-lent character of coordination bonds can also be stud-ied. Fig. 4 reveal the changes of electron density between [CoF6]3- and [Co(CN)6]3- after forming the com-plexes from the neutral atoms. It could be seen from the electron density diagram (Fig. 4a) that when Co and F form coordination bonds, electron densities decreased among atoms (blue contour lines) while sharp increases were observed in space around the atoms, suggesting ionic natures of Co - F bonds. In con-trast, entirely different phenomenon was observed in [Co(CN)6]3- (Fig. 4b). There were significant increases of electron density between C and Co atoms, suggesting covalent nature of Co-C bonds. Thus, from [CoF6]3- to [Co(CN)6]3-, the character of coordination bonds chang-es from mainly ionic to mainly covalent.
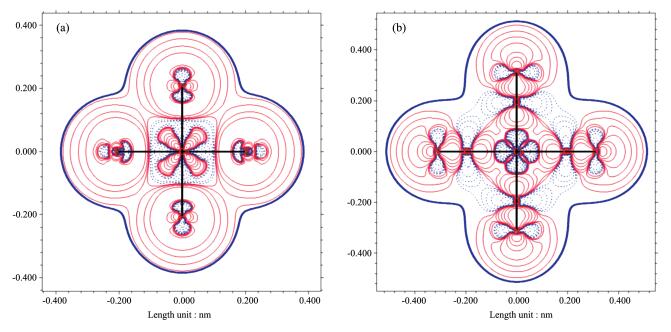
Dotted bule lines and solid red lines indicate the decrease or increase of the electron density after forming coordinating bonds; Solid blue lines indicate the van der Waals surfaces of complexes
Another violation of EN principle of the above complexes is the preferred charge for the central atoms is enormously influenced by the type of coordination atoms. This result reveals that the chemical environ-ment has significant impact on charges of the central atoms. It appears that the charge of the central metal ion decreases even to negative charge with decreasing electronegativities of coordination atoms, as shown in Table 1 and Fig. 1. It is found that when the electroneg-ativity of the coordination atom decreases, electron would not be pulled to the coordination atom but be given to the central atom causing it having a negative charge. NBO analysis revealed that the occupancy of Co-C was 1.90, making it a single bond with bond order of 0.95. The high positive charge and high nega-tive charge of Co3+ in [CoF6]3- and [Co(CN)6]3-, respec-tively, were reflected in their frontier oribital digrams and electron density diagrams (Fig. 3 and 4), showing low electron density and high electron density, respec-tively.
It is also interesting to discover that the negative charge of F, O in H2O and N in NH3 did not decreased by following the trend of electronegativity values of these element, but increasing slightly with N in NH3 bearing the highest charge (Fig. 1), which is probably because H atoms attached to O and N have also trans-ferred some electrons to the coordination atoms, indi-cating they can have small influence to the central metal ions. The overall charges of the neutral ligands were less than 0.5 (Table 1). The NH3 ligand bore more positive charge (0.418) than that of H2O (0.327), consis-tent with the fact that NH3 is a better electron donor ligand than H2O. The CN- bore much less charges than -1, but only -0.273 (Table 1), indicating large amounts of electrons has been donated to Co3+, making Co be-come negative charged (-1.360). F- bore close to -1 charge (-0.802) (Table 1), consistent with the fact that F has the largest electronegativity among all elements except for noble - gas elements. Lone-pair electrons of F- also repel the d electrons of Co, which is a destabi-lizing factor. As a result, the crystal field splitting energy was significantly lower than that of [Co(CN)6]3+ as shown in Fig. 5. The ionic nature of Co -L bonds decreases from F- to C in CN- since the NBO charge of Co decreases as shown in Table 1 and their covalent nature of the Co-L bond increases, meaning better over-laps of related atomic orbitals and shorter Co-L bonds (Fig. 1).
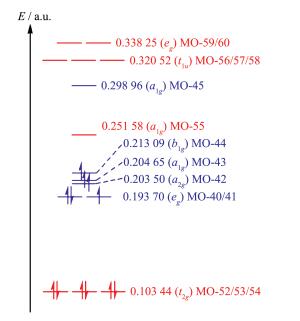
1 a.u.=1 Hartree=2 625.5 kJ·mol-1
Since H2O is a weak field ligand, we calculated all compounds in high-spin states except for Co3+ (Table 2). Calculation results show a decline in NBO charge from 1.758 for Sc3+ to 1.274 for Cr3+, then an increase from Cr3+ to Fe3+ (1.436) followed a decrease to 1.361 for high spin Co3+ (Table 2 and Fig. 6~7). Inter-estingly, there was not even one central metal ion in these hydrates having a charge within -1 to +1. Thus, these examples do not support the EN principle. As the electronegativity values of the central atom increas-es, the corresponding NBO charges generally decreases (Fig. 6). The reason is probably that the less electroneg-ativity value the element has, the more positive charge it tends to have. This is not the case for the range from Cr to Co, suggesting other factors might have effect on the charge of the central atom.
下载:
导出CSV
| NBO charge | Electronegativity of coordination atoms | Bond length (M-O)/nm | r/pmb | |||||
| M | M | -Oa | H | Allen | Pauling | Allred-Rochow | ||
| Sc | 1.785 | 0.973 | 0.203 | 1.19 | 1.36 | 1.20 | 0.216 | 74.5 |
| Ti | 1.539 | 0.935 | 0.243 | 1.38 | 1.54 | 1.32 | 0.210 | 67.0 |
| V | 1.371 | 0.910 | 0.271 | 1.53 | 1.63 | 1.45 | 0.205 | 64.0 |
| Cr | 1.274 | 0.899 | 0.289 | 1.65 | 1.66 | 1.56 | 0.201 | 61.5 |
| Mn | 1.307 | 0.885 | 0.323 | 1.75 | 1.55 | 1.60 | 0.197 | 64.5 |
| Fe | 1.436 | 0.931 | 0.261 | 1.80 | 1.83 | 1.64 | 0.205 | 64.5 |
| CoHc | 1.361 | 0.914 | 0.274 | 1.84 | 1.88 | 1.70 | 0.202 | 61.0 |
| CoLc | 1.034 | 0.865 | 0.327 | 1.84 | 1.88 | 1.70 | 0.192 | 61.0 |
| a Charge of O divided by-1; b Radii data of M3+ from the reference[51]; c CoH or CoL stands for Co in high-spin or low-spin [Co(H2O)6]3+. | ||||||||
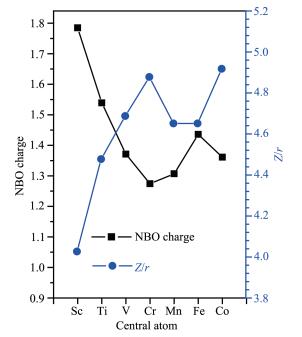
Ionic potential values of M3+ (Z/r with Z=3) were calculated and given in Fig. 7, which have a much bet-ter correlation with NBO charges than electronegativity. The trend is when the ionic potential of the central atom increases, its NBO charge decreases. The reason for the above trend is when ionic potential is high, the M3+ ion would attract more electrons to itself, reducing its positive charge.
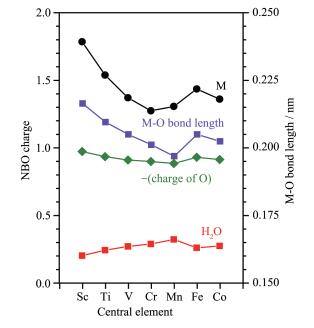
Trends of NBO charges of oxygen atoms and M-O bond lengths (Fig. 8) in [M(H2O)6]3+ correlate well with that of the NBO charges of M. While the charge of M decreases, the absolute charge of O decreases. While the absolute NBO charges of M and O decrease, the covalent nature of the M- O bond increases. This means there are more overlaps between M and O's orbitals and a shorter bond length. Similarly, Co-L bond lengths decrease as the NBO charges of M decreases in the cases of [CoL6] (Fig. 2). When the absolute charge of O decreases, the overall charge of the coordinated H2O increases, implying the charge of H remains constant. The calculated charges of H were 0.587~0.589, 0.589~ 0.590, 0.590~0.591, 0.594, 0.584~0.604, 0.596, 0.596, 0.593~0.594 for M=Sc, Ti, V, Cr, Mn, Fe, Co (LS), Co(HS), respectively. They are very close with a maximum difference of 0.020.
The charges of O was in a very narrow range (±0.1), which means that the radius of O might be roughly the same from Sc to Co. Thus, the M - O bond length would depend on the radius of M3+. This is supported by the reported experimental radius values of M3+ given in Table 1. M- L bond length difference of the adjacent [M(H2O)6]3+ is very similar to the radius difference of the adjacent M3+ except for Mn3+, suggest-ing the radius of Mn3+ might not be accurate as also suggested by Fig. 7. It should be smaller than 64.5 pm[51]. The radius of Mn3+ reported in CRC handbook was 58 pm[52], which gives an improvement of the corre-lation in Fig. 7 and a perfect fit to the calculated Mn-O bond distance. Since radius of Cr3+ (61.5 pm) is 3 pm greater than that of Mn3+ (58 pm), the Mn-O bond distance should be 3 pm less than that of Cr-O, which is 0.198 nm, in agreement with the calculated value of 0.197 nm.
We calculated both the low spin ground state and high spin state of [Co(H2O)6]3+ (Table 2). These states are both thermally accessible and low spin state was reported as the ground state (previous section). NBO charge of Co in low-spin [Co(H2O)6]3+ (1.034) was small-er than that (1.361) of high-spin [Co(H2O)6]3+ (Table 2), and the Co - O bond length of low-spin [Co(H2O)6]3+ (0.192 nm) was shorter than that (0.202 nm) of high - spin [Co(H2O)6]3+.
Parts of the unit cell of α-A2O3 (Fig. S1) were selected and terminated by H2 O or O- to create two compounds, Al3 and Al6. The optimized geometry of two structures and calculated NBO charges are given in Fig. 9 and Table 3. The values were very similar to those obtained by optimizing the structures at B3LYP/6 - 31G(d) level of DFT and NBO analysis at B3LYP/6 - 31G(d, p) or B3LYP/6 -31++G(d, p) level of DFT (Table S1 and S2). The NBO charges of Al in Al3 cluster were 6 - coordinated and in a narrow range (2.039~2.046). Expanding the cluster to Al6 did not change greatly the charges of Al, and they are in a range of 2.084~2.127, slightly larger than those of Al3, except for Al13, which had a charge of 2.039, about 0.10 smaller than the charges of other Al atoms. This Al atom is coordinated with three O2-, and two μ2-O2- atoms is very close to Al (0.172 9~0.173 0 nm), indicating these two Al -O bonds have significant covalent nature, reducing the charge of the Al atom. In contrast, the Al10-μ2-O2- distance was 0.182 9 nm. Charges of Al in Al3 and Al6 fell in very narrow range (2.0~2.1), depending on which atom (ele-ment type and formal charge) it directly connects with, but being not so affected by its coordination number or the type of the atoms two - bonds away. These observa-tions enable us to predict the charge of Al in α-Al2O3. It should be around 2.1±0.1. Real space charge density analysis based on the first - principle, self-consistent orthogonalized linear combination of atomic orbitals (OLCAO) calculations in the local density approximation gave an effective charge formula from Al23+2.63O-1.75 to Al2+2.75O3-1.83 [53]. The experimental values based on common X -ray diffraction data, on the other hand, show that the effective charges on A1 and O were 1.32(5) and -0.88(8), respectively[54]. These numbers are not reliable and tend to underestimate the true val-ues as later discovered by others due to noise, thermal smearing, systematic errors and the X-ray distinction effect[55-56]. Up to date, reliable experimental values for α-Al2O3have not been obtained.
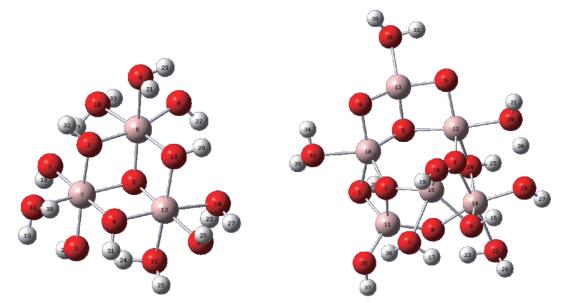
H in white, O in red, Al in pink color
下载:
导出CSV
| Cluster | Al3 | Al6 | |||
| Metal | Atom label, NBO charge, coordination (Ligands) | Atom label, NBO charge, coordination (Ligands) | |||
| Al | 6, 2.039, 6c-Al (1μ1-H2O, 2 μ1-OH-, 2μ2-OH-, 1 μ3-O2-)a | 10, 2.084, 5c-Al (1 μ1-H2O, 1 μ2-OH-, 1 μ2-O2-, 2 μ3-O2-) | |||
| 7, 2.041, 6c-Al (2 μ1-H2O, 1 μ1-OH-, 2 μ2-OH-, 1 μ3-O2-) | 11, 2.113, 4c-Al (1 μ1-OH-, 1 μ2-OH-, 1 μ2-O2-, 1 μ3-O2-) | ||||
| 12, 2.046, 6c-Al (2 μ1-H2O, 1 μ1-OH-, 2μ2-OH-, 1 μ3-O2-) | 12, 2.097, 5c-Al (1 μ1-OH-, 1 μ2-OH-, 1 μ2-O2-, 2 μ3-O2-) | ||||
| 13, 2.039, 4c-Al (1 μ1-H2O, 2 μ2-O2-, 1μ3-O2-) | |||||
| 14, 2.060, 6c-Al (2 μ1-H2O, 2 μ2-OH-, 1 μ2-O2-, 1 μ3-O2-) | |||||
| 15, 2.127, 4c-Al (1 μ1 -H2O, 1 μ2-OH-, 2 μ3-O2-) | |||||
| Ligand | NBO charge of O and (H)b | Sumc | NBO charge of O and (H)b | Sumc | |
| H2O | μ1- | 1, -1.197 (17, 0.540; 18, 0.496) | -0.161 | 7, -1.005 (17, 0.532; 36, 0.540) | 0.067 |
| 9, -0.977 (21, 0.531; 29, 0.543) | 0.097 | 22, -1.010 (23, 0.550; 28, 0.515) | 0.055 | ||
| 11, -0.966 (19, 0.530; 30, 0.537) | 0.101 | 26, -1.201 (27, 0.501; 30, 0.540) | -0.160 | ||
| 14, -0.993 (23, 0.531; 27, 0.541) | 0.079 | 31, -0.996 (32, 0.567; 38, 0.547) | 0.118 | ||
| 15, -1.036 (24, 0.540; 25, 0.522) | 0.026 | 33, -0.988 (34, 0.556; 39, 0.541) | 0.109 | ||
| O- | μ1- | 3, -1.216 (20, 0.497) | -0.719 | 18, -1.222 (19, 0.498) | -0.724 |
| 8, -1.256 (22, 0.499) | -0.757 | 20, -1.044 (21, 0.532) | -0.512 | ||
| 10, -1.037 (33, 0.530) | -0.507 | 35, -1.219 (37, 0.507) | -0.712 | ||
| 16, -1.253 (26, 0.501) | -0.752 | ||||
| μ2- | 2, -1.211 (32, 0.523) | -0.688 | 1, -1.217 (29, 0.541) | -0.676 | |
| 4, -1.204 (31, 0.504) | -0.700 | 9, -1.227 (16, 0.524) | -0.703 | ||
| 13, -1.205 (28, 0.511) | -0.694 | 24, -1.197 (25, 0.514) | -0.683 | ||
| O2- | μ2- | No μ2-O2- in this cluster | 4, -1.432 | ||
| 6, -1.433 | |||||
| 8, -1.475 | |||||
| μ3- | 5, -1.449 | 2, -1.454 | |||
| 3, -1.476 | |||||
| 5, -1.430 | |||||
| a Numbers of each kind of ligands coordinated with the aluminum atoms were given in parentheses, and 6c-Al stands for hexa-coordinated aluminum atom while 5c- and 4c- stand for penta- and tetra-coordinated; b NBO charges of hydrogen atoms connected to the oxygen atoms are given in parentheses; c Sum of NBO charges of O and H in each ligand. | |||||
The charges of O in H2O, OH- and O2- in Al3 were -1.197~ -0.966, -1.256~ - 1.037 and -1.449, respec-tively; for Al6, they were -1.201~ -0.988, - 1.227~ -1.044, - 1.476~ -1.430, respectively. The actual charges of O were not in huge difference from H2O (-1.0~-1.2) to OH- (-1.0~-1.2) and O2- (- 1.4). The smaller or greater ones are due to hydrogen bonding. For example, the charge of O10 of a OH- in Al3 is -1.037, smaller than those of other OH-. This is be-cause of O1-H17…O10 hydrogen bonding (O1-H17 0.153 6 nm, O10-H17 0.103 4 nm, ∠O1- H17…O10= 162°), making O10 also resembles O in H2O, which causes a relative smaller charge.
In summary, the charges of O in OH-, O2- and Al in Al3 and Al6 all exceed 1.0, contradicting to the EN principle.
We find that the EN principle is not applicable to complexes or compounds which contains atoms having high formal charges. [M(H2O)6]3+ (M=Sc, Ti, V, Cr, Mn, Fe, Co), [Co(Ⅲ) L6] (L=F-, H2O, NH3, CN-), Al3O(OH)7 (H2O)5 and Al6O6(OH)6(H2O)5, which contain high for-mal oxidation number atoms, all violates this principle by having atomic charges exceeding +1~ -1. In addi-tion, we find, for compounds having the same charge, that the preferred charge for the central atom is (1) not so influenced by its coordination numbers; (2) not de-termined by its own electronegativity, but can be enor-mously influenced by which elements and their formal charges of the directly coordinated atoms and not so influenced by the atoms two bonds away. Many other examples of compounds having high formal charge at-oms violates the EN principle. For example, Kuroiwa et al. obtained +1.1 for Pb, +2.4 for Ti and -1.4 for O1 and -1.0 for O2 in PbTiO3, which were determined by the MEM (maximum entropy method)/Rietveld analysis using synchrotron-radiation powder data[57]. More calcu-lations and experimental works need to be done to give a better statement for the EN principle.
We predict that the charge of Al in α-Al2O3 is 2.1± 0.1 based on cluster calculations. Reliable experimental charges have not been obtained due to technical prob-lems[55-56], but can be obtained based on high quality X-ray diffraction data obtained by using a synchrotron source and electron diffraction data as done by Zuo et al[55].
In addition, we have the following discoveries. (1) Despite the differences of coordination numbers and ligand type, charges of Al atoms in Al3 and Al6 fell in a very narrow range (2.0~2.1). The actual charges of O atoms in Al3 and Al6 are not in huge difference from H2O (-1.0~-1.2) to OH- (-1.0~- 1.2) to O2- (- 1.4). The smaller or greater ones are due to hydrogen bonding.(2)The charges of Co3+ in [CoF6]3- and [Co(CN)6]3- were found to be 1.639 and -1.360, respectively. (3) When the ionic potential of the central atom increases, its charge would decrease. The M-L bond would have higher covalent nature, and the M-L bond would be shorter as shown in [M(H2O)6]3+ complexes. (4) The spin state of Co3+ strongly affect its charge, which was found to be 1.034 for low - spin state and 1.361 for high spin state. The Co - O bond length of low - spin [Co(H2O)6]3+ (0.192 pm) is shorter than that (0.202 pm) of high-spin [Co(H2O)6]3+. (5) Based on the discovered trend, the radius of Mn3+ should be 58 pm[52] instead of 64.5 pm[51].
Wolynes P G. Ann. Rev. Phys. Chem., 1980, 31:345-376 doi: 10.1146/annurev.pc.31.100180.002021
Martínez M J, Pappalardo R R, Marcos E S, et al. J. Phys. Chem. B, 1998, 102:3272-3282 doi: 10.1021/jp980196d
Keyes T, Napoleon R L. J. Phys. Chem. B, 2011, 115:522-531 doi: 10.1021/jp105595q
Pearson R G.. J. Am. Chem. Soc[J]. , 1963, 85: 3533-3539. doi: 10.1021/ja01076a089
Pearson R G. Science, 1966, 151:172-177 doi: 10.1126/science.151.3707.172
Pearson R G. Chem. Brit., 1967, 3:103-107
Miranda-Quintana R A, Kim T D, Cárdenas C, et al. Theor. Chem. Acc., 2017, 136:135 doi: 10.1007/s00214-017-2167-y
Cartledge G H. J. Am. Chem. Soc., 1928, 50:2855-2863 doi: 10.1021/ja01371a006
Cartledge G H. J. Am. Chem. Soc., 1930, 52:3076-3083 doi: 10.1021/ja01371a006
Feig A L, Panek M, Horrocks W D, et al. Chem. Biol., 1999, 6:801-810 doi: 10.1016/S1074-5521(99)80127-6
Tyler G. Sci. Total Environ., 2004, 329:231-239 doi: 10.1016/j.scitotenv.2004.03.004
Fasfous I I, Yapici T, Murimboh J, et al. Environ. Sci. Technol., 2004, 38:4979-4986 doi: 10.1021/es035427v
徐光宪, 赵学庄.化学学报, 1956, 22:441-446
Sanderson R T. J. Chem. Educ., 1952, 29:539-544 doi: 10.1021/ed029p539
Sanderson R T. J. Chem. Educ., 1954, 31:2-7 doi: 10.1021/ed031p2
Sanderson R T. J. Chem. Educ., 1954, 31:238-245 doi: 10.1021/ed031p238
Pauling L. J. Chem. Soc., 1948, 23:1461-1467
Huheey J E, Keiter E A, Kieter R L. Inorganic Chemistry:Principles of Structure and Reactivity. 4th Ed. New York:HarperCollins College Publishers, 1993:393-394
Crabtree R H. The Organometallic Chemistry of the Transi-tion Metals. 7th Ed. Hoboken:John Wiley & Sons, Inc., 2019:53
Crabtree R H. The Organometallic Chemistry of the Transi-tion Metals. 6th Ed. Hoboken: John Wiley & Sons, Inc., 2014:27-28
Crabtree R H. The Organometallic Chemistry of the Transi-tion Metals. 4th Ed. Hoboken: John Wiley & Sons, Inc. 2005:19-20
García-Lastra J M, Barriuso M T, Aramburu J A, et al. Phys. Rev. B, 2009, 79:241106 doi: 10.1103/PhysRevB.79.241106
Glendening E D, Badenhoop J K, Reed A E, et al. NBO 6.0, University of Wisconsin, Madison, WI, USA, 2013.
Glendening E D, Landis C R, Weinhold F. J. Comput. Chem., 2013, 34:1429-1437 doi: 10.1002/jcc.23266
Reed A E, Weinstock R B, Weinhold F. J. Chem. Phys., 1985, 83:735-746 doi: 10.1063/1.449486
Weinhold F, Landis C R, Glendening E D. Int. Rev. Phys. Chem., 2016, 35:399-440 doi: 10.1080/0144235X.2016.1192262
Wibergand K B, Rablen P R. J. Comput. Chem., 1993, 14:1504-1518 doi: 10.1002/jcc.540141213
Larin A V, Mortier W J, Vercauteren D P. J. Comput. Chem., 2007, 28:1695-1703 doi: 10.1002/jcc.20660
Turnbull D, Wetmore S D, Gerken M. Angew. Chem. Int. Ed., 2019, 58:13035-13038 doi: 10.1002/anie.201906600
Chatterjee K, Dopfer O. Chem. Sci., 2018, 9:2301-2318 doi: 10.1039/C7SC05124G
Yang Y F, Houk K N, Wu Y D. J. Am. Chem. Soc., 2016, 138:6861-6868 doi: 10.1021/jacs.6b03424
Cohen A J, Mori-Sánchez P, Yang W. Chem. Rev., 2012, 112:289-320 doi: 10.1021/cr200107z
Sproviero E M, Gascon J A, McEvoy J P, et al. J. Inorg. Biochem., 2006, 100:786-800 doi: 10.1016/j.jinorgbio.2006.01.017
Crystallography Open Database (No. 1000017). http://www.crystallography.net/
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016.
Sophia P, Andrey Z, Julia S, et al. Cryst. Mater., 2018, 233:35-40 doi: 10.1515/zkri-2016-2021
Deeth R J, Foulis D L, Williams-Hubbard B J. Dalton Trans., 2003, 1:3949-3955 doi: 10.1039/b305868a
Cotton F A, Meyers M D. J. Am. Chem. Soc., 1960, 82:5023-5026 doi: 10.1021/ja01504a002
Allen G C, Warren K D. Struct. Bond., 1971, 9:49-138 doi: 10.1007/BFb0118885
Monajjemi M, Khaleghian M. J. Clust. Sci., 2011, 22:673-692 doi: 10.1007/s10876-011-0414-2
Hepworth M A, Jack K H, Peacock R D, et al. Acta Crystal-logr., 1957, 10:63-69 doi: 10.1107/S0365110X57000158
Babel D. Struct. Bond., 1967, 3:1-87
Friedman H L, Hunt J P, Plane R A, et al. J. Am. Chem. Soc., 1951, 73:4028-4030 doi: 10.1021/ja01152a521
Johnson D A, Sharpe A G. J. Chem. Soc. A, 1966, 1:798-801 doi: 10.1039/j19660000798
Nielsen M T, Moltved K A, Kepp K P. Inorg. Chem., 2018, 57:7914-7924 doi: 10.1021/acs.inorgchem.8b01011
Mortensen S R, Kepp K P. J. Phys. Chem. A, 2015, 119:4041-4050 doi: 10.1021/acs.jpca.5b01626
Johnson D A, Nelson P G. Inorg. Chem., 1999, 38:4949-4955 doi: 10.1021/ic990426i
Winkler J R, Rice S F, Gray H B. Comments Inorg. Chem., 1981, 1:47-51 doi: 10.1080/02603598108078079
Navon G. J. Phys. Chem., 1981, 85:3547-3549 doi: 10.1021/j150624a001
Johnson D A, Nelson P G. J. Chem. Soc. Dalton Trans., 1990, 19:1-4 doi: 10.1039/DT9900000001
Speight J G. Lange's Handbook of Chemistry. 16th Ed. New York: McGRAW-HILL, 2005:1.153
Lide D R. CRC Handbook of Chemistry and Physics, Internet Version 2007. 87th Ed. Boca Raton: Taylor and Francis, 2007: 12.12. http:/www.hbcpnetbase.com
Ching W Y, Xu Y N. J. Am. Ceram. Soc., 1994, 77:404-411 doi: 10.1111/j.1151-2916.1994.tb07008.x
Lewis J, Schwarzenbach D, Flack H D. Acta Crystallogr. A, 1982, 38:733-739 doi: 10.1107/S0567739482001478
Zuo J M, Kim M, O'Keeffe M, et al. Nature, 1999, 401:49-52 doi: 10.1038/43403
Pillet S, Souhassou M, Lecomte C, et al. Acta Crystallogr. Sect. A, 2001, A57:290-303 doi: 10.1107/S0108767300018626
Kuroiwa Y, Aoyagi S, Sawada A, et al. Phys. Rev. Lett., 2001, 87:217601 doi: 10.1103/PhysRevLett.87.217601
Lu T, Chen F W. J. Comput. Chem., 2012, 33:580-592 doi: 10.1002/jcc.22885
Martin F, Zipse H. J. Comput. Chem., 2005, 26:97-105 doi: 10.1002/jcc.20157
Clark A E, Sonnenberg J L, Hay P J, et al. J. Chem. Phys., 2004, 121:2563-2570 doi: 10.1063/1.1766292
卢天, 陈飞武.物理化学学报, 2012, 28(1):1-18 doi: 10.3866/PKU.WHXB201401211

Figure 1 Trend of NBO charge on central cobalt atom, -(charge of coordinated atoms), and each ligand compared with three scale (Pauling's, Allred-Rochow's and Allen's) electronegativity of coordination atoms

Figure 2 Optimized geometry of [Co(CN)6]3- and [CoF6]3-
Co-C: 0.195 0 nm; Co-F2, F4, F5 or F7: 0.205 5 nm; Co-F3 or F6:0.191 4 nm

Figure 3 Selected frontier orbital diagrams of [CoF6]3- (a) and [Co(CN)6]3- (b) drawn by Multiwfn[58] software

Figure 4 Electron density diagrams of [CoF6]3- (a) and [Co(CN)6]3- (b) obtained by subtracting the electron density of the related neutral atoms from the electron density of actual complex using Multiwfn[58] software based on the wavefunction data obtained in the geometry optimization step
Dotted bule lines and solid red lines indicate the decrease or increase of the electron density after forming coordinating bonds; Solid blue lines indicate the van der Waals surfaces of complexes

Figure 5 Energy levels of selected frontier orbitals (shown in Fig. 3) of [CoF6]3- (in blue) and [Co(CN)6]3-(in red)
1 a.u.=1 Hartree=2 625.5 kJ·mol-1

Figure 6 Trend of NBO charges on central transition metal trivalent ions of [M(H2O)6]3+ (High-spin Co3+ data included), compared with three scales (Pauling's, Allred-Rochow's and Allen's) of electronegativity of central transition metal elements


Figure 8 Trend of NBO charges on O in H2O, compared with that of high spin central transition metal trivalent ions

Figure 9 Atomic numbers and NBO charges of optimized structures of Al3 and Al6
H in white, O in red, Al in pink color
Table 1. NBO charges of CoL6(L=F-, H2O, NH3, CN-) and other related data
| NBO charge | Electronegativity of coordination atoms | Bond length (M-O) / nm | |||||
| L | Co | -(coord. atom)* | L | Allen | Pauling | Allred-Rochow | |
| F- | 1.639 | 0.802 | -0.802 | 4.19 | 3.98 | 4.10 | 0.206 |
| H2O | 1.034 | 0.865 | 0.327 | 3.61 | 3.44 | 3.50 | 0.192 |
| NH3 | 0.489 | 0.952 | 0.418 | 3.07 | 3.04 | 3.07 | 0.203 |
| CN- | -1.360 | -0.375 | -0.273 | 2.54 | 2.55 | 2.50 | 0.195 |
| *Charge divided by -1. | |||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. NBO charges of [M(H2O)6]3+ (M=Sr~Co) and related data
| NBO charge | Electronegativity of coordination atoms | Bond length (M-O)/nm | r/pmb | |||||
| M | M | -Oa | H | Allen | Pauling | Allred-Rochow | ||
| Sc | 1.785 | 0.973 | 0.203 | 1.19 | 1.36 | 1.20 | 0.216 | 74.5 |
| Ti | 1.539 | 0.935 | 0.243 | 1.38 | 1.54 | 1.32 | 0.210 | 67.0 |
| V | 1.371 | 0.910 | 0.271 | 1.53 | 1.63 | 1.45 | 0.205 | 64.0 |
| Cr | 1.274 | 0.899 | 0.289 | 1.65 | 1.66 | 1.56 | 0.201 | 61.5 |
| Mn | 1.307 | 0.885 | 0.323 | 1.75 | 1.55 | 1.60 | 0.197 | 64.5 |
| Fe | 1.436 | 0.931 | 0.261 | 1.80 | 1.83 | 1.64 | 0.205 | 64.5 |
| CoHc | 1.361 | 0.914 | 0.274 | 1.84 | 1.88 | 1.70 | 0.202 | 61.0 |
| CoLc | 1.034 | 0.865 | 0.327 | 1.84 | 1.88 | 1.70 | 0.192 | 61.0 |
| a Charge of O divided by-1; b Radii data of M3+ from the reference[51]; c CoH or CoL stands for Co in high-spin or low-spin [Co(H2O)6]3+. | ||||||||
下载: 导出CSV
Table 3. NBO charges of the atoms and ligands of Al3 and Al6
| Cluster | Al3 | Al6 | |||
| Metal | Atom label, NBO charge, coordination (Ligands) | Atom label, NBO charge, coordination (Ligands) | |||
| Al | 6, 2.039, 6c-Al (1μ1-H2O, 2 μ1-OH-, 2μ2-OH-, 1 μ3-O2-)a | 10, 2.084, 5c-Al (1 μ1-H2O, 1 μ2-OH-, 1 μ2-O2-, 2 μ3-O2-) | |||
| 7, 2.041, 6c-Al (2 μ1-H2O, 1 μ1-OH-, 2 μ2-OH-, 1 μ3-O2-) | 11, 2.113, 4c-Al (1 μ1-OH-, 1 μ2-OH-, 1 μ2-O2-, 1 μ3-O2-) | ||||
| 12, 2.046, 6c-Al (2 μ1-H2O, 1 μ1-OH-, 2μ2-OH-, 1 μ3-O2-) | 12, 2.097, 5c-Al (1 μ1-OH-, 1 μ2-OH-, 1 μ2-O2-, 2 μ3-O2-) | ||||
| 13, 2.039, 4c-Al (1 μ1-H2O, 2 μ2-O2-, 1μ3-O2-) | |||||
| 14, 2.060, 6c-Al (2 μ1-H2O, 2 μ2-OH-, 1 μ2-O2-, 1 μ3-O2-) | |||||
| 15, 2.127, 4c-Al (1 μ1 -H2O, 1 μ2-OH-, 2 μ3-O2-) | |||||
| Ligand | NBO charge of O and (H)b | Sumc | NBO charge of O and (H)b | Sumc | |
| H2O | μ1- | 1, -1.197 (17, 0.540; 18, 0.496) | -0.161 | 7, -1.005 (17, 0.532; 36, 0.540) | 0.067 |
| 9, -0.977 (21, 0.531; 29, 0.543) | 0.097 | 22, -1.010 (23, 0.550; 28, 0.515) | 0.055 | ||
| 11, -0.966 (19, 0.530; 30, 0.537) | 0.101 | 26, -1.201 (27, 0.501; 30, 0.540) | -0.160 | ||
| 14, -0.993 (23, 0.531; 27, 0.541) | 0.079 | 31, -0.996 (32, 0.567; 38, 0.547) | 0.118 | ||
| 15, -1.036 (24, 0.540; 25, 0.522) | 0.026 | 33, -0.988 (34, 0.556; 39, 0.541) | 0.109 | ||
| O- | μ1- | 3, -1.216 (20, 0.497) | -0.719 | 18, -1.222 (19, 0.498) | -0.724 |
| 8, -1.256 (22, 0.499) | -0.757 | 20, -1.044 (21, 0.532) | -0.512 | ||
| 10, -1.037 (33, 0.530) | -0.507 | 35, -1.219 (37, 0.507) | -0.712 | ||
| 16, -1.253 (26, 0.501) | -0.752 | ||||
| μ2- | 2, -1.211 (32, 0.523) | -0.688 | 1, -1.217 (29, 0.541) | -0.676 | |
| 4, -1.204 (31, 0.504) | -0.700 | 9, -1.227 (16, 0.524) | -0.703 | ||
| 13, -1.205 (28, 0.511) | -0.694 | 24, -1.197 (25, 0.514) | -0.683 | ||
| O2- | μ2- | No μ2-O2- in this cluster | 4, -1.432 | ||
| 6, -1.433 | |||||
| 8, -1.475 | |||||
| μ3- | 5, -1.449 | 2, -1.454 | |||
| 3, -1.476 | |||||
| 5, -1.430 | |||||
| a Numbers of each kind of ligands coordinated with the aluminum atoms were given in parentheses, and 6c-Al stands for hexa-coordinated aluminum atom while 5c- and 4c- stand for penta- and tetra-coordinated; b NBO charges of hydrogen atoms connected to the oxygen atoms are given in parentheses; c Sum of NBO charges of O and H in each ligand. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们