School of Materials Science and Engineering, Anhui University of Science and Technology, Huainan, Anhui 232001, China
2.
School of Chemistry and Chemical Engineering, Yancheng Institute of Technology, Yancheng, Jiangsu 224051, China
3.
School of Materials Science and Engineering, Yancheng Institute of Technology, Yancheng, Jiangsu 224051, China
4.
School of Materials Science and Engineering, Jiangsu University, Zhenjiang, Jiangsu 212013, China
5.
Key Laboratory for Advanced Technology in Environmental Protection of Jiangsu Province, Yancheng Institute of Technology, Yancheng, Jiangsu 224051, China
Received Date:
01 April 2020 Revised Date:
07 May 2020 Available Online:
10 September 2020
Abstract:
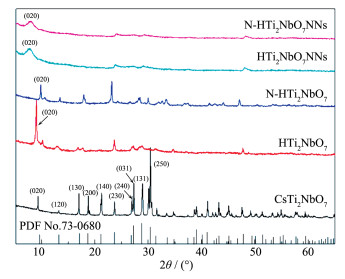
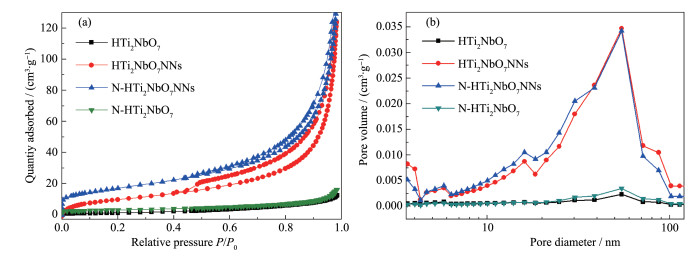
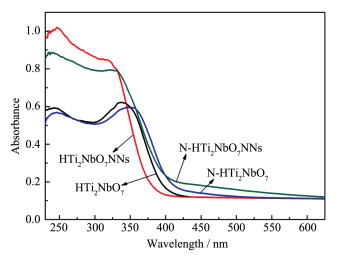
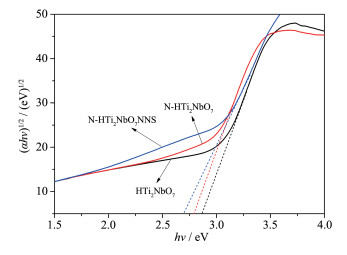
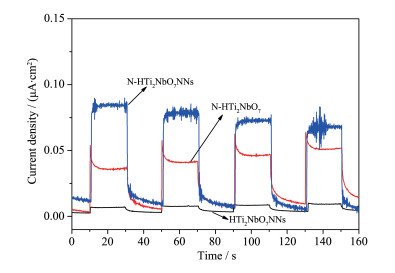
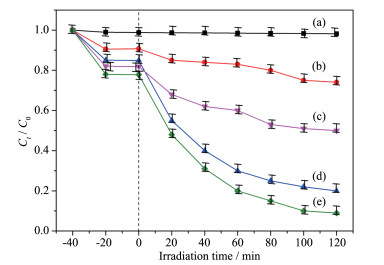
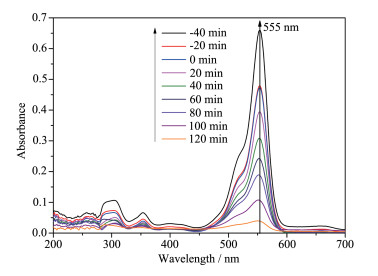
Firstly, layered CsTi2NbO7 was prepared by high temperature solid-state method, and then layered HTi2NbO7 was obtained by a proton-exchange reaction treated with HNO3 solution. Secondly, the obtained layered HTi2NbO7 was well dispersed in tetrabutylammonium hydroxide (TBAOH) solution in order to prepare HTi2NbO7 nanosheets by exfoliation reaction. Thirdly, the as-prepared HTi2NbO7 nanosheets were dried, and then mixed with urea. Finally, nitrogen-doped HTi2NbO7 nanosheet photocatalyst was successfully synthesized by heating the above mixture. The as-prepared samples were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), high resolution transmission electron microscopy (HRTEM), X-ray photoelectron spectroscopy (XPS), Ultraviolet-Visible diffuse reflectance absorption spectrum (UV-Vis DRS) as well as N2 adsorption-desorption measurements to characterize the crystal structure, morphology, specific surface area, pore distribution and light absorption capacity of as-prepared samples in detail. It was found that the doped nitrogen resulted in an intrinsic narrowing band-gap so as to widen light response region, and the doped nitrogen atoms were mainly located at the interstitial position of Ti2NbO7- sheets and chemically bonded with hydrogen ions. Moreover, compared with N-doped HTi2NbO7, N-doped HTi2NbO7 nanosheets had a larger specific surface area and richer mesoporous structure due to the relatively loose and irregular arrangement of the titanium niobate nanosheets. Therefore, the N-doped HTi2NbO7 nanosheets exhibited higher visible-light photocatalytic activity for degradation of RhB than that for N-doped layered HTi2NbO7.
a BET (Brunauer-Emmett-Teller) specific surface area calculated from the linear part of BET plot; b Total pore volume taken from the volume of N2 adsorbed at p/p0=0.98; c Average pore diameter was estimated from the BJH formula
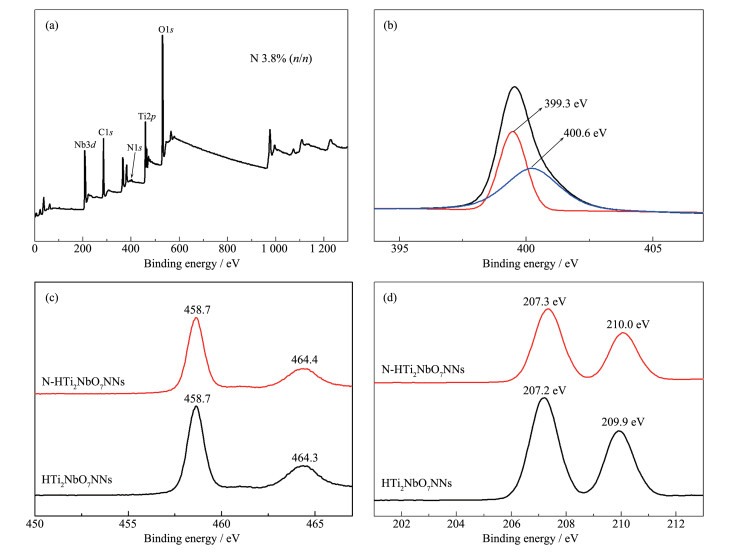
Figure 4.
(a) XPS survey and (b) high resolution of N1s spectra of N-HTi2NbO7NNs samples; (c) High resolution XPS spectra of Ti2p and (d) Nb3d of HTi2NbO7NNs and N-HTi2NbO7NNs samples
Figure 4
(a) XPS survey and (b) high resolution of N1s spectra of N-HTi2NbO7NNs samples; (c) High resolution XPS spectra of Ti2p and (d) Nb3d of HTi2NbO7NNs and N-HTi2NbO7NNs samples
Table 1.
Specific surface area (SBET), pore volume (Vt) and average pore size (D) of samples
Sample
SBETa / (m2·g-1)
Vtb / (cm3·g-1)
Dc / nm
HTi2NbO7
3
0.018
-
N-HTi2NbO7
5
0.021
-
HTi2NbO7NNs
41
0.188
33.6
N-HTi2NbO7NNs
63
0.208
29.8
a BET (Brunauer-Emmett-Teller) specific surface area calculated from the linear part of BET plot; b Total pore volume taken from the volume of N2 adsorbed at p/p0=0.98; c Average pore diameter was estimated from the BJH formula

 下载:
下载:


 下载:
下载:












