引用本文:
王文杰, 陈玉霞, 梁玲, 曾波, 谈洁, 袁荃. 基于微生物体系合成无机纳米材料的研究进展[J]. 无机化学学报,
2020, 36(5): 777-794.
doi:
10.11862/CJIC.2020.108

Citation: WANG Wen-Jie, CHEN Yu-Xia, LIANG Ling, ZENG Bo, TAN Jie, YUAN Quan. Recent Advances in Microbial Biosynthesis of Inorganic Nanomaterials[J]. Chinese Journal of Inorganic Chemistry, 2020, 36(5): 777-794. doi: 10.11862/CJIC.2020.108
Citation: WANG Wen-Jie, CHEN Yu-Xia, LIANG Ling, ZENG Bo, TAN Jie, YUAN Quan. Recent Advances in Microbial Biosynthesis of Inorganic Nanomaterials[J]. Chinese Journal of Inorganic Chemistry, 2020, 36(5): 777-794. doi: 10.11862/CJIC.2020.108
基于微生物体系合成无机纳米材料的研究进展
English
Recent Advances in Microbial Biosynthesis of Inorganic Nanomaterials
Abstract:
Inorganic nanomaterials are gradually playing important roles in a myriad of fields such as energy and biomedicine. Therefore, the development of various protocols for the synthesis of inorganic nanomaterials has attracted widespread attention in the last decades. Biomineralization common in nature endows living organisms the ability to synthesize various inorganic nanomaterials with marvellous structures and diverse features. Inorganic nanomaterials synthesized by microbes are eco-friendly, low-cost, and biocompatible, thus becoming an important issue in nanomaterial science. In this review, we provide a brief overview of the recent research efforts on the microbial biosynthesis of inorganic nanomaterials and their mechanisms, affecting factors as well as potential applications. In addition, the current challenges and future perspective of microbial biosynthesis of inorganic nanomaterials have also been discussed.
-
Key words:
- biosynthesis
- / inorganic nanomaterial
- / microbe
- / nanoparticle
- / synthetic method
-
0. 引言
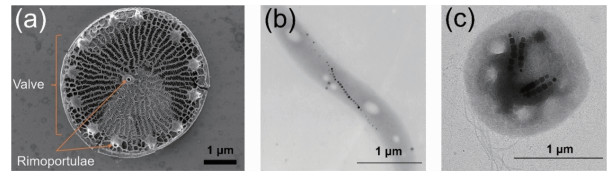
在30多亿年漫长的进化历程中,生物体已经能够产生具有独特和精致结构的矿物质,如珊瑚石、牙齿、骨骼和贝壳等等[1-2]。这一过程被称为生物矿化,即生物体利用有机基质(酶、蛋白质、脂类、多糖等)对无机离子进行精确调控,最终沉淀成具有复杂结构的纯无机或者无机有机混合材料[3-5]。生物矿化是生物体维系生存的一种行为[3],在细菌、真菌等微生物中广为存在。微生物作为地球上最古老的一类生命体,具有体形极小、结构简单、繁殖速度快等特点[6],能够利用生物矿化过程合成出各种微小的无机材料。例如,为了在呼吸、集光的同时形成“盔甲”,硅藻等海洋生物可以让SiO2在细胞表面快速成核、固定,形成具有微米到毫米层级结构的硅质外骨骼,并在细胞死亡后也能保持完好(图 1a)[7-9];一些厌氧性细菌被称为趋磁细菌(magnetotactic bacteria),它们能够在细胞内合成形态各异的Fe3O4或Fe3S4磁性纳米晶体并呈链状分布(磁小体链),从而根据地磁场进行定向移动来摆脱富养环境(图 1(b,c))[10-11];有的嗜金属微生物群能在污水区域大量繁殖,因为它们能够将水溶性的重金属富集并以多种形状结晶析出来抵御金属毒性[12-13]。微生物作为分布最为广泛的一类生物[6, 14-15],常常暴露于极端的自然环境之下,它们需要合成各种晶形或非晶形微纳尺寸的无机材料来维持自己的生存[14, 16-17],这使它们成为了合成无机纳米材料的天然工厂。
图 1
在过去的二十年间,纳米材料正在成为物理、化学、材料等领域的研究重点。纳米材料因尺寸极小(1~100 nm),具有表面效应、量子尺寸效应等特性而表现出与其宏观材料完全不同的物理化学性能[13, 18-19]。其中无机纳米材料在催化、传感、环境、医学等领域扮演重要角色[13],并且其通常由无机物前体通过物理或化学方法制备而成[20-23]。不同于传统方法,自然界中的微生物已天然具有将无机离子合成为含金、银、钙、硅、铁等无机纳米材料的能力[24]。1980年,Beveridge等[25]首次发现枯草芽孢杆菌(B. subtilis)能够将溶液中的Au3+固定还原成粒径不到50 nm的Au纳米颗粒(AuNPs)。1999年,Klaus等[26]发现硝酸银溶液会使施氏假单胞菌AG259(P. stutzeri AG259)的周质空间内形成高度分散、具有多种晶形的Ag纳米颗粒(AgNPs)。微生物塑造纳米材料的现象在上世纪末引起了纳米科学家们的广泛关注,这给无机纳米材料的合成带来了新的启发。过去几十年间,研究者们已经利用微生物或者微生物提取物合成了Au、Ag、Pd、TiO2、CdS等一系列无机纳米材料并阐明了部分合成机理和影响因素[13],发现了微生物体系合成的无机纳米材料具有诸多优点。首先,微生物体系合成无机纳米材料的过程与微生物的生长、酶的催化等生物过程是同时进行的,保证了合成过程可以在室温和中性pH条件下发生,环保而且成本较低。其次,微生物对无机纳米材料表面进行的生物修饰也使得材料的生物相容性、分散性和稳定性较好[27-28]。上述优势使得微生物合成技术有望成为合成无机纳米材料的重要手段。
利用微生物体系合成无机纳米材料正成为一个新兴的、具有广阔前景以及多学科交叉的研究领域,并且有望在未来进行商业化应用。近年来报道了许多关于微生物体系合成无机纳米材料的综述,如Gahlawat和Choudhury[23]从合成机理的角度综述了微生物合成金属纳米材料的研究进展;李清彪和杨大鹏课题组[29]介绍了了植物、微生物等多种生物合成金属纳米材料的研究;Dahoumane等[30]综述了藻类合成无机纳米材料的进展;张春阳课题组[31]总结了近年来微生物体系和仿生体系合成半导体量子点(QDs)及其生物医学应用的研究;庞代文课题组[32]综述了微生物活细胞合成无机纳米材料的生物应用研究进展。但是目前关于微生物体系合成不同种类无机纳米材料的机理、影响因素,以及这些材料在多个领域应用的综述还较少。为了较为全面地了解基于微生物体系合成无机纳米材料的相关研究,我们将从细菌、真菌、酵母等多种微生物体系合成的金属和非金属等不同种类无机纳米材料进行系统的综述,并从其合成机理、影响因素及其在催化、生物医学等领域的应用等方面进行了阐述,最后我们也对微生物体系合成无机纳米材料目前存在的问题以及未来的发展进行了分析和展望。
1. 微生物合成无机纳米材料的机理
不同的微生物有着不同的无机纳米材料合成机制。总体来讲,微生物的合成机制来源于3种生物活动过程:微生物对极端环境压力的抵抗、对无机营养的利用和对能量代谢的需求[13, 33]。微生物对极端环境压力的抵抗能力来自于其对金属离子的耐毒性机制,包括对金属氧化态的改变、细胞壁吸附、多肽或多糖的螯合、射流系统等等[33-35];微生物对无机营养的需求指的是微生物主动利用无机离子合成自身的特殊结构,如硅藻主动吸收硅酸盐合成外骨骼;微生物的能量代谢需求则是微生物将无机基质用于生物体代谢过程以获得还原性的当量,并用于生物合成或能量储存[13, 33]。微生物体系合成无机纳米材料的过程是微生物分泌的多种酶或蛋白质等生物活性分子协同作用的结果,并且不同的合成过程会利用到不同的生物活性分子中。其中常见的参与合成的生物活性分子包括还原型辅酶Ⅰ(NADH)[36]、还原型辅酶Ⅱ(NADPH)[37-38]、硝酸还原酶[34, 37-39]、植物螯合肽[13, 34, 38]、氢化酶[40]、多糖[24, 34, 41]、谷胱甘肽[34, 42]等等。细菌、真菌体系通常利用还原性酶的作用[24],酵母体系倾向于利用植物螯合肽的螯合过程[13, 38]。大部分微生物体系合成无机纳米材料的过程包括成核、生长和稳定3个步骤,因此其往往能合成具有有序晶体结构的纳米材料。
微生物合成无机纳米材料的机理是极其复杂且多样的,即便对于同种微生物也存在多种机制来合成无机纳米材料,这增加了对合成机理研究的难度。其中较为常见的是生物还原(Bioreduction)所主导的合成机制,它是指高价态金属离子被微生物体系还原成低价态金属离子或金属单质的过程[23, 29, 34],此外其他种类纳米材料的合成机制还受多种复杂代谢过程的控制。理解微生物合成无机纳米材料的机理是控制其产品性质的关键,以下将对生物还原和其他合成机理分别进行讨论,以进一步了解各种生物分子在合成过程中的作用。
1.1 生物还原主导的合成机理
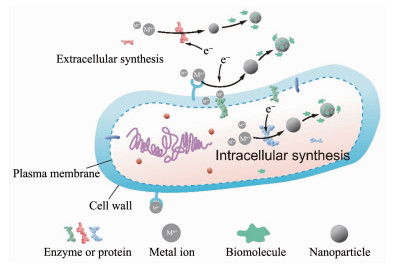
生物还原过程几乎存在于所有微生物合成无机纳米材料的过程中,它体现了微生物对于金属毒性的抵抗作用。生物还原主导的合成是指在还原性酶或者蛋白质等活性物质的催化下,一些生物分子(谷胱甘肽[43]、NADH[36]、NADPH[37]、蛋白质等)作为电子供体还原金属离子,生成水溶性更差或毒性更低的单质或低价态金属。生物还原合成的场所分为细胞内和细胞外两种,这取决于参与反应的生物活性分子的分布位置(图 2)。细胞内合成时,金属离子被膜转运系统输送到细胞质后被生物活性分子所还原[39, 42, 44-46]。细胞外合成时有2种情况,第1种为微生物向胞外分泌的生物活性分子直接在胞外还原金属离子[34, 46-48],含有微生物分泌物的培养液上清液同样也能还原金属离子[39, 42, 44];第2种是在细胞表面合成,细胞壁表面的羧基、磷酸基团等先通过静电引力吸附金属离子(少部分情况通过共价结合),然后细胞壁、细胞膜上的生物活性分子还原金属离子[13-14, 33-34, 44, 49]。还原开始后,生物分子中吸引金属离子的基团会帮助金属颗粒快速成核、生长[14, 50]。最后为了增加材料的稳定性,微生物会利用蛋白质、多糖等生物分子作为天然的包覆剂和稳定剂对颗粒进行包裹,由于静电斥力和空间位阻的作用,纳米颗粒长时间内不易团聚[39, 51-53]。
图 2
 图 2. 无机纳米材料的生物还原合成示意图Figure 2. Biosynthesis of inorganic nanomaterials mediated by bioreduction
图 2. 无机纳米材料的生物还原合成示意图Figure 2. Biosynthesis of inorganic nanomaterials mediated by bioreductionBiosynthesis of inorganic nanomaterials mediated by bioreduction can be classified into extracellular synthesis and intracellular synthesis; Metal ions are reduced followed by nucleation and growth of nanoparticles; Dispersed nanoparticles are obtained after stabilization by biomolecules
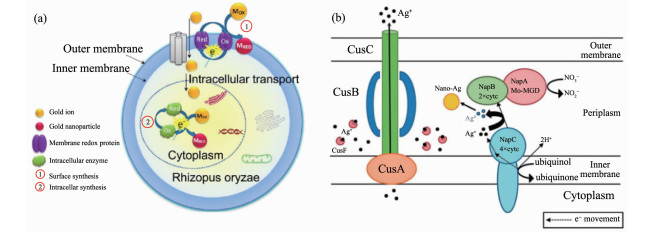
金属离子的生物还原过程往往是多种场所同时进行的。Das等[44]认为米根霉(R. oryzae)合成AuNPs时,一部分Au3+在细胞表面被蛋白质还原成AuNPs,此外另一部分Au3+渗透进入细胞质与蛋白质共价结合并被金属还原酶还原成AuNPs(图 3a)。许多细胞外的生物还原过程还涉及到细胞利用复杂的电子传递途径进行呼吸作用的过程[23, 53]。香港大学支志明的团队[49]证明了细胞色素c在大肠杆菌合成AgNPs中起到了电子传递的重要作用,他们认为耐银性大肠杆菌与AgNO3溶液反应时,细胞氧化呼吸底物的同时细胞色素c的亚基NapC介导了周质空间内银离子的还原(图 3b)。
图 3
 Figure 3. (a) Proposed mechanism of AuNPs biosynthesis by fungi R. oryzae[44]; (b) Proposed mechanism of AgNPs biosynthesis mediated by c-type cytochrome subunit NapC in E. coli strain[49]
Figure 3. (a) Proposed mechanism of AuNPs biosynthesis by fungi R. oryzae[44]; (b) Proposed mechanism of AgNPs biosynthesis mediated by c-type cytochrome subunit NapC in E. coli strain[49]MOX: oxidized Au3+ ion; MRED: reduced metallic Au; Red: reduced protein or enzyme; Ox: oxidized protein or enzyme
1.2 其他合成机理
对于一些处于稳定氧化态的无机离子,微生物难以通过生物还原过程将其沉淀,此时微生物需要利用更为复杂的机制来合成无机纳米材料。大部分非生物还原主导的合成过程同样具有成核、生长和稳定的过程,并且在胞内或胞外进行,但是目前很多机理尚未完全阐明。一些氧化物的合成来自于微生物介导的水解过程,如硅藻合成SiO2可能与细胞壁上硅蛋白参与的硅酸盐水解有关[8],TiO2的微生物合成同样来自于生物诱导的TiF62-的水解[54-55],病毒特有的表面结构还可以作为模板通过水解来合成氧化物纳米材料涂层[56]。趋磁细菌合成Fe3O4纳米颗粒的机理有多种解释,其中一种认为:趋磁细菌先在胞内形成具有碱性环境的磁小体囊泡,然后细胞膜上的Fe3+被部分还原形成Fe2+及铁蛋白并进入囊泡当中,Fe3+和Fe2+在囊泡上膜蛋白的作用下快速共沉淀,成核、生长产生Fe3O4颗粒[10, 21]。Fe3S4纳米颗粒的合成过程则更为复杂,其合成过程中有更多的无机相存在,目前还研究得较少[10]。CdS、PbS等含剧毒重金属的半导体材料的合成往往与多肽等物质的螯合作用有关[13]。其中植物螯合肽和金属硫蛋白是微生物中非常常见的稳定重金属离子的螯合剂,这是因为植物螯合肽中的谷胱甘肽、金属硫蛋白中的半胱氨酸基团可以有效螯合金属离子[13, 43]。这些生物源的螯合剂会在毒性条件下过表达以快速解毒,如酵母中的谷胱甘肽和植物螯合肽会高表达将Cd2+、Pb2+富集[13, 42],然后半胱氨酸提供硫源以合成CdS、PbS纳米颗粒[57-58]。还有一些微生物合成的无机纳米材料是自身代谢过程得到的副产品,如细菌表面的S层蛋白可以促进方解石等细粒矿物的成核[14],硫酸盐还原菌自身的硫代谢途径促进了ZnS纳米颗粒的形成[59]。总之,微生物合成无机纳米材料的机理十分复杂,目前很多生物分子在合成当中的作用尚未被完全理解,还需要进一步的研究了解微生物合成无机纳米材料的分子和基因机制。
2. 影响微生物合成的因素
合成具有理想的成分、尺寸和形貌特征的无机纳米材料一直是纳米技术面临的挑战。在物理或化学方法中,影响合成的因素往往比较明确,而微生物合成无机纳米材料的性质则受各种复杂因素的影响,导致了尺寸、形状、合成速率相差很大[30]。影响合成效果的因素有很多,包括微生物的种类、pH值、温度、金属离子浓度、菌种生长期、培养基成分、孵育时间、微生物或蛋白浓度、离子强度、是否搅拌等等[30, 39, 46, 60-63]。下面我们主要阐述了最重要的前4种因素[64]及其对微生物体系合成金属纳米材料的影响。
2.1 微生物的种类
微生物是种类极其繁多的生物群体,通常包括细菌、病毒、真菌等肉眼难以观察到的小型生物。目前被用于合成无机纳米材料的微生物有细菌、真菌、放线菌、病毒、酵母和藻类等[42, 60, 65-66],图 4展示了几种微生物和它们合成的无机纳米材料。由于不同的微生物具有不同的基因、蛋白和代谢过程,进行生物矿化的功能分子不同,因而它们合成无机纳米材料的能力也不同[23]。
图 4

细菌因为结构简单、生长快、容易培养[14]而最早被用于合成无机纳米材料[43]。细菌由于表面的净负电性电荷和S层蛋白[14, 68]而很容易吸附金属离子并提供成核位点。细菌中大肠杆菌的基因编辑技术较为成熟,通过基因编辑可以增强其合成无机纳米材料的某些效果[69-71]。真菌是合成无机纳米材料的主力军,它们相比于其他微生物具有许多突出的优点:能承受更多的外界机械压力,可以在无机基质上生长,生长面积大,对金属离子的摄入量高,向外分泌的酶和蛋白量比其他微生物多,因而合成效率很高[34, 42, 51, 65]。放线菌作为细菌的一种同样生长繁殖快速,但放线菌同时又具有许多重要的真菌特征,如菌丝比表面积大、胞外分泌蛋白量多[34],尽管放线菌具有诸多优点,但目前对其合成能力的探索还较少。酵母是一种能够积累大量剧毒金属的单细胞真菌[13, 34, 42]。酵母的解毒能力主要来源于其富含的谷胱甘肽、金属硫蛋白和植物螯合肽等生物分子,其中植物螯合肽可以高效地螯合Cd2+等剧毒重金属离子[13, 34, 43],因此酵母很适合用于合成CdS、CdSe等纳米材料[34, 72]。植物螯合肽也有利于纳米颗粒的稳定和分散[13],因而酵母能很容易地在胞内合成小粒径的半导体量子点[58, 67]。酵母也有基因编辑的潜力[73],这允许其像重组大肠杆菌一样调控合成效果。藻类通常是水生的光合自养生物[30],其中有部分生物(如蓝藻)属于微生物的范畴。藻类特有的光合系统电子传递链也能起到合成无机纳米材料的重要作用[53],并且藻类等水生生物适应恶劣环境的能力很强[74],因此藻类是很有潜力的“纳米工厂”[30, 34]。病毒很擅长利用生物还原制备无机纳米材料[23, 29, 66],因为病毒衣壳蛋白独特的拓扑结构和残基有利于金属离子的结合和还原[29, 75]。病毒衣壳还可以通过基因工程进行表面修饰,成为一种组装具有复杂结构的纳米材料的模板[76],从而为金属离子提供均匀、精确的结合位点,并对金属离子进行选择性还原[23, 56, 75]。病毒与其他微生物在生物结构和合成无机纳米材料的过程方面差别过大,Lee等[56]对病毒合成无机纳米材料进行了详细的阐述。微生物合成无机纳米材料的过程存在着高度的生物控制,产物的晶体形态和成分很大程度上取决于微生物的特性[77],对此我们在合成不同形貌及大小的无机纳米材料时应选择合适的生物资源。
2.2 pH值
大量研究表明pH值能显著影响微生物体系合成金属纳米材料的形貌大小和合成速率,如荚膜红假单胞菌(R. capsulata)在pH=4时胞外合成了大于200 nm的金纳米片,而在pH=7时主要合成10~20 nm大小的纳米颗粒[36];轮枝孢菌(V. luteoalbum)在pH=5和7时合成AuNPs的粒径要远大于pH=3和9时[78];云芝(C. versicolor)蛋白在碱性条件下合成AgNPs反应速率比中性更快,反应时间从72 h减少到1 h[79];节杆菌属(Arthrobacter sp.)胞外多糖在pH=5以下便失去了合成AgNPs的能力,而在pH=7到8之间能快速还原Ag+ [41];一些微生物及其提取物的合成速率还在某一特定pH值下达到最大值[80-81]。因此,pH值被认为是影响微生物合成的一个重要因素[82]。
pH值对不同微生物合成的金属纳米材料会产生不同的效果。其中一种关于pH值影响微生物合成的解释认为,质子浓度会影响生物活性分子的总电荷,进而影响它们对金属离子的吸附积累量[83]和对晶体生长的控制能力[82]。以AuNPs的合成过程为例,pH值较低时质子浓度很大,容易使参与还原过程的氨基、巯基、羧基等基团带更多正电荷,导致了其还原能力和还原速率的下降[33, 36, 48],但同时生物活性分子也更容易吸附Au3+与生物分子形成的负电性络合物或者AuCl4-,最终聚集形成大块的金纳米片[33, 48];pH值较高时则生物活性分子的还原能力增强、还原速率增加,但带负电性的Au3+络合物或AuCl4-与生物分子的结合能力变差,易于形成热力学上更稳定的小粒径、单分散的球形AuNPs[36, 62]。过于酸、碱性的环境不仅不利于还原过程的进行,还可能会引起材料表面电荷的不稳定,导致团聚现象发生[41, 62]。此外,pH值还可能影响酶和蛋白的活性、细胞携带的电荷、细胞膜的通透性等因素,最终得到不同的合成效果,并可能因此产生合成过程的最适pH值。
2.3 温度
微生物体系作为一个复杂的化学反应体系,必然受温度控制。一般来说,随着温度的升高,微生物体系合成金属纳米材料的速率会加快。例如,摩根氏菌属(M. psychrotolerans)合成AgNPs的速率在4~25 ℃的温度范围内随温度不断提高[84];一些耐温范围更广的微生物或微生物提取物,它们合成金属纳米材料的速率也会随着温度的升高而增加[78, 85-86]。李玉梅等[41]探究了不同温度孵育下Arthrobacter sp.胞外多糖合成AgNPs的速率。当孵育温度为70 ℃时,10 min内就可以观察到AgNPs的形成;当温度升至80 ℃时,合成过程为5 min;而在90 ℃时,合成过程则不到2 min。也有研究表明微生物体系合成一些金属纳米材料的过程存在最快反应温度,如南极假单胞菌(P. Antarctica)为22 ℃[80],瘿青霉(P. Fellutanum)为10~20 ℃[81]。温度的改变同样能影响材料的形貌,如M. psychrotolerans在20 ℃时主要合成球形AgNPs,而在4和25 ℃时主要合成多边形银纳米片[84];V. luteoalbum在50 ℃时会生成大的球形或多种其他形状的金纳米材料,而在25 ℃时仅有小粒径颗粒形成[78]。
温度对微生物体系合成金属纳米材料的影响机制目前还不够清楚。一般认为,一定范围内温度的升高加速了金属离子的细胞内积累和还原过程[46, 60],有利于颗粒表面二次还原的发生从而形成大颗粒[86],但反应物的消耗速度和材料的成核速度的加快有时反而会导致形成小粒径颗粒[63, 85],具体的影响效果还需要视合成途径而定。温度过高会导致材料表面生物分子变性以及酶的失活[62-63],不仅不会继续提高合成效率,还会不利于其稳定分散,导致团聚现象更加严重[87]。
2.4 金属离子浓度
金属离子是微生物合成金属纳米材料的原料离子。微生物虽然具有一定的金属耐受性,但大量的金属离子对于微生物结构的破坏是不可避免的。随着金属离子浓度的增大,更多的金属离子参与到合成过程中,导致最初微生物合成速率显著增加,但过高的金属离子浓度会使微生物死亡或生物分子失活,最终导致合成效率急剧下降[60, 81]。目前对于金属离子浓度与金属纳米材料形貌、大小的关系也有许多人进行了研究。V. luteoalbum被报道在HAuCl4浓度较小时(250和500 mg·L-1)合成20 nm左右的纳米颗粒,当HAuCl4浓度增至2 500 mg·L-1时,合成出来的则是200 nm左右形状不规则的金纳米片[78];Riddin的团队[88]利用硫酸盐还原菌群的蛋白进行Pt纳米颗粒合成时,发现H2PtCl6与蛋白的质量浓度比值的改变会明显影响颗粒形貌,当比值为0.7:1时容易形成大块的不规则颗粒并且团聚现象严重,比值为2:1时则会形成粒径较小、分散性较好的方块状颗粒;东南大学顾宁课题组[89]发现R. capsulata在HAuCl4物质的量浓度较低时只会产生10~20 nm的球形AuNPs,而当HAuCl4浓度较高时则会形成具有网状结构的金纳米线,他们认为这和包覆剂(参与合成和稳定过程的蛋白质等生物分子)与HAuCl4的浓度比值以及奥斯特瓦尔德成熟(Ostwald ripening)过程有关;Das等[61]对真菌R. oryzae提取物合成AuNPs的影响因素进行了深入研究(图 5),他们同样认为金属纳米颗粒的大小和形状受包覆剂与金属离子质量浓度的比值控制,这些参与合成和稳定过程的蛋白在晶体上的吸附水平会随着金属离子浓度的改变而改变,从而诱导产生了不同形状的晶体。
图 5

3. 微生物合成的无机纳米材料
无机纳米材料因其独特的光学、电学、磁学和热学等性能受到了广泛关注,不同无机纳米材料的性能也各不相同。为了满足各种应用的需要,科学家们对微生物体系合成无机纳米材料的能力进行了广泛探索,成功地诱导了多种微生物从无机离子前体合成出不同的无机纳米材料,并通过多种手段表征了这些材料的理化性质以及探究了其在不同应用领域的发展前景。下面将从微生物体系所合成的各种无机纳米材料的角度进行综述,并在表 1中归纳了一些微生物合成的无机纳米材料。
表 1
 表 1 一些微生物合成的无机纳米材料及其性质Table 1. Some inorganic nanomaterials synthesized by microbes and their properties
表 1 一些微生物合成的无机纳米材料及其性质Table 1. Some inorganic nanomaterials synthesized by microbes and their properties 下载:
导出CSV
下载:
导出CSV
Microorganism Product Size / nm Shape Application Ref. Pycnoporus sanguineus Au 10~40 Spherical Electrocatalyst [90] Rhizopus oryzae Au 10 Spherical Water purification [91] Rhizopus oryzae Au Diverse sizes Diverse shapes — [61] Plectonema boryanum Au 25 Cubic — [92] Fusarium oxysporum Ag 1.6 Spherical Antibacterial [93] Rhizopus oryzae Ag 15 Spherical Water purification [94] Phaenerochaete chrysosporium Ag 50~200 Pyramidal — [95] Pseudomonas stutzeri Ag 200 Diverse shapes — [26] Shewanella oneidensis Cu 20~50 Spherical Catalyst [96] Enterobacter sp. Hg 2~5 Spherical — [97] Shewanella sp. Pd 13 — Electrocatalyst [98] Sulfate-reducing bacteria Pt 50~300 Rectangular — [88] Shewanella oneidensis Pd-Au alloy 4.95 — Electrocatalyst [99] Magnetospirillum magneticum Fe3O4 20~30 Cubic — [100] Shewanella oneidensis Pd/Fe3O4, Au/Fe3O4, PdAu/Fe3O4 3~15 — Catalyst [101] Actinobacter sp. γ-Fe2O3, Fe3S4 5~7 — — [102] Fusarium oxysporum TiO2, SiO2 5~15 Irregular or spherical — [54] Aspergillus flavus TiO2 12~15 — Photosynthesis [103] Bacillus licheniformis ZnO 200~1000 Flower Photocatalyst [104] Lactic acid Bacillus Sb2O3 3~12 Cubic — [105] Moorella thermoacetica CdS < 10 — Photosynthesis [106] Schizosaccharomyces pombe CdS 2~2.5 Hexagonal Diode [107] Sulfate-reducing bacteria ZnS 2~5 Spherical — [59] Yeast CdTe 2~3.6 — Bio-imaging [67] Recombinant Escherichia coli CdSeZn, PrGd, CdCs, FeCo etc. Diverse sizes Atypical shapes — [70] Yeast CaCO3 4~6 — Drug delivery [108] Bacillus subtilis Se 50~400 Spherical Biosensor [109] Sulfate-reducing bacteria Graphene — — Electrocatalyst [110] —: Not reported 3.1 金属纳米材料
3.1.1 金和银纳米材料
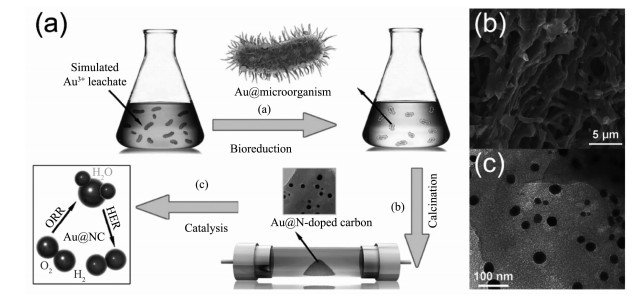
由于合成过程简单,过去几十年间金和银纳米材料被视作研究微生物合成无机纳米材料的简单模型而得到了广泛的研究[28, 39, 48]。1980年,Beveridge等[25]最早发现细菌B. subtilis能够合成AuNPs,而后2001年Mukherjee等首次利用真菌合成了AuNPs[111]和AgNPs[112],目前人们已经能够运用多种微生物资源合成具有复杂结构的金、银纳米材料,如顾宁课题组[89]报道了用R. capsulata合成出金纳米线,最近Sreedharan等[64]还成功地用B. subtilis合成出了具有较高比表面积的金纳米花。微生物体系合成的金、银纳米材料不仅在形貌上高度可控,还因为在光、电、抗菌等性能方面表现优异而具有一定的应用潜力。早期微生物体系合成的金和银纳米材料常应用于医疗、环境领域,如Durán等[93]用尖孢镰刀菌(F. oxysporum)制备出的AgNPs对棉衣进行抗菌化处理,一些真菌合成的金和银纳米材料还被用于促进伤口愈合[113]、消毒净水[91, 94]等。此外,Joerger等[114]率先以细菌P. stutzeri AG259细胞作为碳基质,并将细胞碳基质和细菌合成的AgNPs以薄膜形式沉积在铝基上合成出具有典型金属陶瓷性能的陶瓷-Ag复合材料。这种将微生物同时作为无机纳米材料的还原载体和应用载体的方法可以有效绕过无机纳米材料与微生物分离这一繁琐的步骤[29],并能在一定程度上提高其理化性质,为后来微生物合成金属-碳材料的研究提供了新颖的思路。2016年,加州大学圣克鲁兹分校陈少伟和华南理工大学周伟家等团队[90]利用细菌膜作为电催化载体来改善微生物合成AuNPs的电催化性能。他们首先在血红密孔菌(P. sanguineus)表面和内部合成出了AuNPs,该AuNPs紧密吸附在经过Ar气氛高温煅烧后成为了氮掺杂碳基材料(NC)的细胞膜上,最终组成了负载AuNPs的氮掺杂碳基复合材料(Au@NC)(图 6)。C、N元素的引入显著增强了AuNPs的电化学性能,Au与NC之间存在大量的电荷迁移过程,因此Au@NC表现出对析氢反应(HER)及氧还原反应(ORR)高效的电催化活性。除了微生物膜,其他纳米材料也能同时成为金、银纳米材料的还原和应用载体以增强微生物合成金和银纳米材料的性能。安徽大学石先阳课题组[115]利用奥奈达希瓦氏菌(S. oneidensis)在硝酸改性的多壁碳纳米管(MWNTs)上原位合成了高度分散的AgNPs。改性后的MWNTs因具有大量成核位点而容易促进S. oneidensis合成高度分散的AgNPs,MWNTs优异的导电性能还显著增强了Ag催化降解4-硝基苯酚的能力。除了与纳米材料结合,一些微生物合成的AuNPs还会很容易与生物分子本身组成偶联物而增强AuNPs的吸附或催化性能[116],因为AuNPs表面的生物修饰作用,微生物合成的AuNPs还表现出比对照组AuNPs更强的表面增强拉曼(SERS)效应[52]。由于微生物体系合成的金和银纳米材料具有形貌可控、性能优异、合成过程简单等特点,在过去几十年间微生物合成技术已经发展成为合成金、银纳米材料的一种重要手段,未来将有可能在大规模生产方面实现重要突破。
图 6
 图 6. (a) Au@NC的合成过程示意图[90]; (b) Au@NC的SEM图中的纤维形态的Au@NC及亮点状的AuNPs[90]; (c) Au@NC的TEM图中的AuNPs负载于NC上且粒径为10~40 nm[90]Figure 6. (a) Preparation of N-doped carbon-supported AuNPs (Au@NC)[90]; (b)Fibrous morphology of Au@NC and bright Au dots in SEM image[90]; (c) AuNPs with sizes ranging from 10 to 40 nm loaded on N-doped carbon (NC) in TEM image[90]
图 6. (a) Au@NC的合成过程示意图[90]; (b) Au@NC的SEM图中的纤维形态的Au@NC及亮点状的AuNPs[90]; (c) Au@NC的TEM图中的AuNPs负载于NC上且粒径为10~40 nm[90]Figure 6. (a) Preparation of N-doped carbon-supported AuNPs (Au@NC)[90]; (b)Fibrous morphology of Au@NC and bright Au dots in SEM image[90]; (c) AuNPs with sizes ranging from 10 to 40 nm loaded on N-doped carbon (NC) in TEM image[90]AuNPs were synthesized on the cell surface and in the cell, then Au/microbe composite was further calcined to prepare Au@NC as an electrocatalyst for hydrogen evolution reaction andn oxygen evolution reaction
3.1.2 其他金属纳米材料
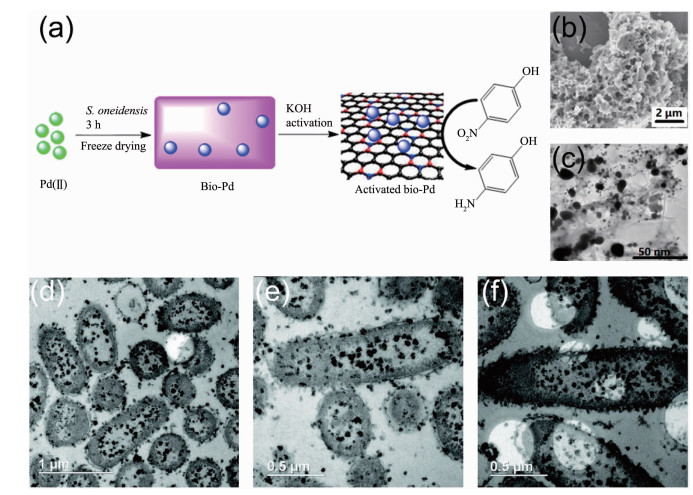
受到微生物合成金、银纳米材料的启发,人们探索了微生物合成其他金属纳米材料的能力。其中Pd、Pt两种金属单质具有出色的稳定性和催化性能,所以开发微生物合成Pd、Pt纳米材料的方法显得尤为重要。Lloyd的团队[117]最早尝试了以丙酮酸、甲酸或H2等外加物质为电子供体,利用微生物来回收废液中的Pd2+并得到了Pd纳米颗粒,几年后Lengke的团队[118]又用微生物合成出了Pt纳米颗粒。由于微生物对金属离子的吸附作用,Pd、Pt纳米颗粒往往会沉淀在细胞表面并被细胞膜有效分散和稳定,其催化活性得到了提高[119-120]。基于这一点,中国科学技术大学俞汉青课题组[98, 121]进一步提出用高温碳化方法由S. oneidensis合成杂原子掺杂的Pd-C介孔纳米材料以提高Pd纳米颗粒的催化性能和导电性能。他们首先利用细菌S. oneidensis还原PdCl42-,在细胞表面合成了生物源的Pd纳米颗粒(Bio-Pd),接着向Pd与微生物的混合溶液中加入KOH高温活化数小时,KOH高温脱水形成的K2O与微生物膜反应产生了内部微孔和缺陷,最终合成出负载Pd纳米颗粒的杂原子掺杂的介孔碳材料(图 7a)。导电性的介孔碳材料具有较高的比表面积且Pd在介孔碳上分散良好(图 7(b~c)),因而该Pd-C介孔纳米材料表现出良好的催化降解4-硝基苯酚的能力和氧还原反应电催化活性。石先阳课题组[122]还利用S. oneidensis在TiO2纳米管上原位制备出了Pd-TiO2复合光催化剂,进一步扩展了微生物合成Pd纳米材料在光催化领域的应用。除了Pd、Pt,微生物合成其他金属纳米材料的能力也得到了初步探索,如S. oneidensis合成Cu纳米颗粒(CuNPs)(图 7(d~f))[96]、肠杆菌属(Enterobacter sp.)合成Hg纳米颗粒[97]、乳酸菌合成Ti纳米颗粒[123]、多种细菌合成La等纳米晶体[124]等。这些方法合成的金属纳米材料具有良好的分散性和稳定性,对后续实现微生物合成多种金属纳米材料具有重要的借鉴意义。
图 7
 图 7. (a) 由S. oneidensis合成负载Pd纳米颗粒的杂原子掺杂介孔碳材料示意图[121]; (b) 400 ℃高温活化后S. oneidensis细胞和Pd纳米颗粒的SEM图[121]; (c) 400 ℃高温活化后S. oneidensis细胞和Pd纳米颗粒的TEM图[121]; (d~f) S. oneidensis在细胞内大量合成的CuNPs的TEM图[96]Figure 7. (a) Preparation of heteroatom-doped porous carbon materials with Pd nanoparticles synthesized by S. oneidensis[121]; (b) SEM image of Pd nanoparticles and S. oneidensis cells after activation at 400 ℃[121]; (c) TEM image of Pd nanoparticles and S. oneidensis cells after activation at 400 ℃[121]; (d~f) TEM images of large amounts of CuNPs synthesized in S. oneidensis cytoplasm[96]
图 7. (a) 由S. oneidensis合成负载Pd纳米颗粒的杂原子掺杂介孔碳材料示意图[121]; (b) 400 ℃高温活化后S. oneidensis细胞和Pd纳米颗粒的SEM图[121]; (c) 400 ℃高温活化后S. oneidensis细胞和Pd纳米颗粒的TEM图[121]; (d~f) S. oneidensis在细胞内大量合成的CuNPs的TEM图[96]Figure 7. (a) Preparation of heteroatom-doped porous carbon materials with Pd nanoparticles synthesized by S. oneidensis[121]; (b) SEM image of Pd nanoparticles and S. oneidensis cells after activation at 400 ℃[121]; (c) TEM image of Pd nanoparticles and S. oneidensis cells after activation at 400 ℃[121]; (d~f) TEM images of large amounts of CuNPs synthesized in S. oneidensis cytoplasm[96]3.1.3 双金属纳米材料
由于协同效应,双金属纳米材料往往在某些方面比金属单质性能更突出[99, 125],因此近年来出现了许多关于微生物合成双金属纳米材料的研究。Nair和Pradeep[126]首次提出用乳酸菌同时还原金和银前体在细胞内合成Au-Ag合金纳米材料,进行了微生物合成双金属纳米材料的重要尝试,后来Senapati等[127]进一步采用真菌胞外合成Au-Ag合金纳米材料并改善了其分散性,这一系列的研究使得微生物合成双金属纳米材料的能力引起了广泛关注。微生物体系合成双金属纳米材料的合成过程简单、分散性较好,具有用于制备电极材料的潜力,如武汉大学胡成国的团队[128]将酵母胞外合成的Au-Ag合金纳米颗粒用于修饰电化学传感器并将其灵敏度提高了5倍。但是非导电性的微生物膜容易吸附合金纳米材料、高温碳化处理会导致颗粒团聚等问题往往限制了其在电化学等领域的应用。对此华中农业大学韩鹤友和中科院城市环境研究所赵峰的团队[99]提出利用细菌S. oneidensis基于氧化石墨烯(GO)和高温碳化过程合成合金-碳阳极材料以解决这些问题。他们首先在细菌S. oneidensis表面先后还原PdCl4-、AuCl4-得到了微生物膜负载的Pd-Au合金材料,然后在微生物膜-Pd-Au材料表面进一步涂覆GO,最后通过水热反应制备出了具有三维多孔结构的杂原子掺杂的Pd-Au合金/还原氧化石墨烯(rGO)复合材料。高温碳化和包裹rGO后的细胞膜导电性显著提升,并且预先进行GO的包裹避免了高温碳化时颗粒聚集的问题,提高了合金材料的比表面积。因此,这种合金纳米材料和碳纳米材料的组装方法将大大提升合金纳米材料的电学性能。安徽大学程园园等[129]在催化应用方面也采取了类似的策略,但是他们没有进行高温碳化过程,而是用S. oneidensis一步还原合成了Pd-Ag/rGO复合材料,并将其用于催化降解污染物,充分展示了细菌S. oneidensis在合成金属-碳纳米材料方面的巨大潜力。总之,S. oneidensis等微生物因具有强大的生物还原能力为合成双金属纳米材料提供了巨大探索空间,从而成为微生物体系合成无机纳米材料在催化、电化学领域应用的重要生物资源。
3.2 金属氧化物和金属硫化物纳米材料
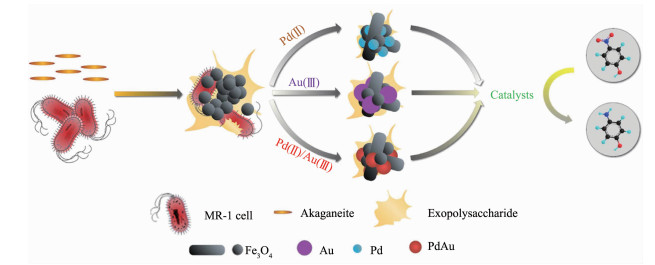
由于合成机制复杂,人们对微生物体系合成金属氧化物和金属硫化物纳米材料的探索经历了漫长的过程。趋磁细菌是最早被发现可以合成金属氧化物和金属硫化物纳米材料的微生物之一,1975年Blakemore[130]在Science上首次展示的这种能在细胞内合成链状磁性纳米颗粒的微生物引起了当时多个领域的关注。趋磁细菌能利用Fe3+在细胞内合成具有长方形、子弹形、菱形等多种晶形的Fe3O4、Fe3S4、γ-Fe2O3等磁性纳米颗粒[10, 77],因此其一直被当作生物矿化过程的标准模型而得到了广泛研究[10]。尽管其基因层面的合成机理尚不明确,但人们已经从中学习并利用其他多种微生物合成了Fe3O4、Fe3S4、Co3O4、FeS2等磁性纳米材料[10, 43, 131],如Bharde等[132]利用真菌胞外分泌蛋白使铁前体水解形成方块状Fe3O4纳米颗粒;Byrne的团队[133]尝试了利用硫还原地杆菌(G. sulfurreducens)合成Zn置换的磁铁矿纳米颗粒。相比于非磁性纳米材料,由微生物合成的磁性纳米材料具有许多独特的性质[134]。首先,磁性纳米材料更容易从反应体系中回收利用。大连理工大学柳广飞的团队[101]利用S. oneidensis MR-1先在胞外合成了Fe3O4纳米颗粒,然后在Fe3O4表面进一步通过生物还原过程合成了Pd/Fe3O4、Au/Fe3O4和PdAu/Fe3O4纳米复合材料(图 8),该复合材料不仅能有效催化降解有机污染物,Fe3O4纳米颗粒的存在还使得该催化剂可以通过磁场回收;Coker等[135]用铁还原菌同样得到了Fe3O4纳米颗粒负载的Pd催化剂;Watson等[136-137]认为硫酸盐还原菌产生的FeS具有吸附水中重金属离子的能力,并且FeS能够通过磁场从水中分离而实现水的净化。其次,一些趋磁细菌合成的磁性纳米材料具有高度可控的形貌特征、良好的生物相容性和超顺磁性,因而在生物医学领域具有广阔的应用前景[10, 57],例如Lisy的团队[138]将趋磁细菌中磁螺菌(M. gryphiswaldense)合成的磁性纳米材料作为造影剂,用于巨噬细胞的磁共振成像(MRI);Matsunaga等[21]还综述了趋磁细菌等微生物合成的磁性纳米材料在生物医学领域的应用,包括药物递送、DNA提取、免疫分析等。这些成果都表明对合成磁性纳米材料的微生物的研究具有重要的意义。
图 8

不同于趋磁细菌,硅藻可以利用细胞壁表面的蛋白质将海水中的硅酸盐水解而产生紧密排布的SiO2材料。受微生物这种强大水解活性的启发,研究者们可以由微生物合成许多金属氧化物,如真菌F. oxysporum被报道通过生物诱导的水解作用由TiF62-和SiF62-溶液合成TiO2和SiO2纳米颗粒[54]以及由ZrF62-溶液合成ZrO2纳米颗粒[55];乳酸菌被报道由Sb3+合成了Sb2O3纳米颗粒[105]。除了上述氧化物外,UO2[139]、Ag2O[140]、CeO2[141]、Bi2O3[142]、CuO[143]等纳米材料均能由微生物以多种涉及水解或氧化过程的机制合成。在这些氧化物当中,一些半导体纳米材料因具有出色的光电性能而被用作光催化剂。例如,Raliya等[103]利用黄曲霉(A. flavus)胞外合成了粒径只有几纳米的TiO2纳米颗粒,他们将TiO2喷洒到植物叶面后有效促进了植物的生长,这表明微生物合成的TiO2纳米颗粒具有良好的光催化性能和生物相容性;Tripathi的团队[104]还利用地衣芽孢杆菌(B. licheniformis)合成了ZnO纳米花,并表明B. licheniformis合成的ZnO具有较强的光催化降解有机污染物的性能。
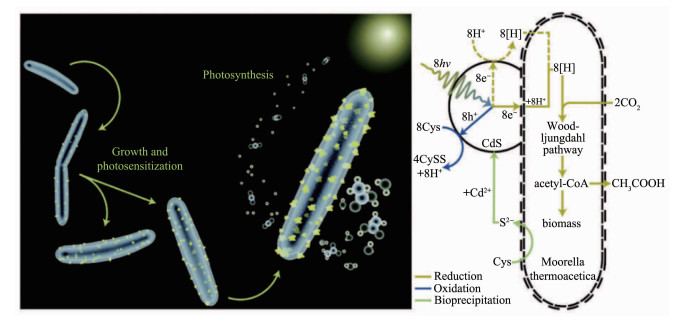
与上述合成氧化物的机制有所不同,许多微生物合成金属硫化物纳米材料的能力是来自于对重金属胁迫的抵抗。1989年,Dameron等[144]最早发现酵母可以在螯合肽的作用下由Cd2+溶液合成CdS QDs;Cunningham和Lundie[145]接着在1993年指出半胱氨酸和CdCl2能使大肠杆菌表面沉淀出CdS颗粒。一般认为,微生物为了避免重金属离子的毒性作用,他们会利用半胱氨酸等含硫分子诱导重金属离子沉淀而解除毒性。研究者们已经发现酵母在这方面表现出色,它们利用细胞内富含的多肽将重金属离子螯合并在液泡当中隔离[13, 32],进而利用半胱氨酸合成多种硫化物纳米晶体。由于酵母细胞的解毒能力出色,且酵母合成的无机纳米材料往往尺寸较小而均一,它们一直在金属硫化物纳米材料的合成当中扮演着重要的角色[13, 31, 34],如酵母被报道合成CdS[107]、PbS[146]、ZnS[147]等多种纳米材料。除了酵母,许多微生物也广泛具有合成CdS[148]、ZnS[59]、PbS[148-149]、Ag2S[26, 150]、HgS[148, 151]、Bi2S3[152]、CuS[153]等金属硫化物的能力,但是合成机制不尽相同。无论是哪种机制,微生物合成的许多金属硫化物纳米材料具有良好的晶体结构和较小的尺寸,故这些材料很好地保持了半导体的性能[13]。杨培东课题组[106]以半胱氨酸作为硫源在非光合作用的CO2还原细菌热醋穆尔氏菌(M. thermoacetica)细胞表面合成了CdS纳米颗粒,并通过CdS半导体纳米颗粒与细菌Wood-Ljungdahl固碳途径的结合实现了非光合细菌对太阳能的有效利用。半导体CdS在光照下跃迁产生的大量电子可以参与到非光合细菌M. thermoacetica还原CO2的电子链当中,使其进行光合作用产生乙酸(图 9),并且量子产率达到了85%左右。Kowshik的团队[107]还将粟酒裂殖酵母(S. pombe)合成的CdS纳米晶用于制备二极管装置,由于颗粒表面生物修饰的作用,该二极管具有工作电压低,正向电流大等特点。这些研究表明微生物将有望成为合成半导体纳米材料的小型工厂,未来应当进行更多的探索。
图 9

3.3 其他金属化合物纳米材料
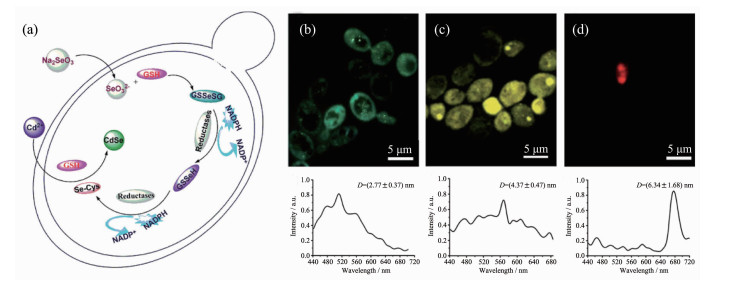
除了硫化物半导体量子点外,微生物还具备合成其他多种量子点的能力。例如,武汉大学庞代文课题组[32]提出将酵母内Se的还原代谢途径和Cd2+离子的解毒途径相耦合用于合成CdSe QDs,他们首先利用酵母细胞中的谷胱甘肽及相关还原性酶将Na2SeO3还原成Se-Cys复合物,然后复合物在谷胱甘肽的作用下与Cd2+结合最终形成CdSe QDs(图 10a)[58]。他们还进一步研究了CdSe QDs的控制合成及发光性质,结果表明随着Cd2+孵育时间的延长,CdSe QDs的直径不断变大,由2.77 nm左右变为6.34 nm左右,CdSe的发光波长也随之红移(图 10(b~d))。基于酵母合成CdSe QDs优异的发光性能,中国药科大学吴盛美和严拯宇的团队[154]进一步利用含有CdSe QDs的酵母细胞作为荧光探针检测水中和血浆中的Cu2+,首次实现了利用活细胞内生物合成的量子点进行离子选择性检测。不同于上述胞内合成量子点的过程,赵东元院士的团队[67]另辟蹊径在酵母胞外合成了CdTe QDs,他们指出胞外合成的CdTe QDs同样具有尺寸可调、荧光发射较强的特点,并且胞外合成的量子点极易回收处理,CdTe QDs在被酵母内吞后还可以实现对酵母细胞的原位生物成像。微生物合成的各种半导体量子点因具有出色的水溶性和生物相容性,在生物医学领域具有广阔的应用前景,对此张春阳课题组[31]有详细的综述。为了进一步提升微生物合成量子点等无机纳米材料的能力,Park等[70]利用基因编辑技术由微生物合成了多种量子点以及其他金属纳米材料。他们将其他物种的植物螯合肽合成酶和金属硫蛋白的基因引入大肠杆菌后,成功地合成出了CdZn、CdSe、CdTe、SeZn、CdSeZn、PrGd、CdCs和FeCo等无机纳米材料。这项研究充分说明了基因编辑技术将成为微生物合成新型无机纳米材料的一把利剑,能进一步丰富微生物合成无机纳米材料的种类。
图 10
 Figure 10. (a) Proposed mechanism of CdSe QDs biosynthesis by yeast[58]; (b~d) In situ laser confocal scanning microscopy photographs and corresponding fluorescence spectra of yeasts after incubation with Cd2+ for (b) 12, (c) 24, and (d) 40 h, respectively[58]
Figure 10. (a) Proposed mechanism of CdSe QDs biosynthesis by yeast[58]; (b~d) In situ laser confocal scanning microscopy photographs and corresponding fluorescence spectra of yeasts after incubation with Cd2+ for (b) 12, (c) 24, and (d) 40 h, respectively[58]GSH: glutathione; Diameters(D) of CdSe QDs increased gradually after incubation with Cd2+ for a period of time; In situ laser confocal scanning microscopy photographs and corresponding fluorescence spectra show that yeasts emitted green fluorescence, yellow fluorescence and red fluorescence after incubation with Cd2+ for 12, 24 and 40 h respectively
微生物合成CaCO3、CaSO4等无机盐的例子在自然界中相当常见。微生物通过生物矿化合成这些矿物质来维持自身的生存,因此利用微生物这种与生俱来的能力可以合成一些无机盐类纳米材料。例如,河南师范大学杨林的团队[108]利用酵母呼吸产生的CO2在生物分子的引导下与溶液中的Ca2+反应在胞内合成了CaCO3纳米颗粒,这种内源性的CaCO3支架可以诱导药物进入细胞,然后作为pH响应的药物递送载体;厦门大学贾立山课题组[155]还利用了毕赤酵母(P. pastoris)提取物合成了Cu掺杂的LaCoO3光催化剂,Cu的掺杂和材料表面的生物修饰使得该催化剂对甲醛溶液制氢反应具有较强的光催化活性。此外,微生物细胞生物矿化的能力还被用于合成细胞外壳。浙江大学唐睿康课题组[156]通过提高酵母细胞表面的电荷密度,在酵母细胞表面原位诱导合成了一层磷酸钙外壳,该外壳不仅保持了细胞的生命活力,还可以在恶劣的外部条件下保护细胞。除了上述无机盐外,微生物还被用于合成SrCO3[157]、BaTiO3[158]、FePO4[50]等多种纳米材料。基于微生物复杂的代谢过程,未来利用微生物合成各种具有复杂成分和结构的无机纳米材料成为了可能。
3.4 其他非金属纳米材料
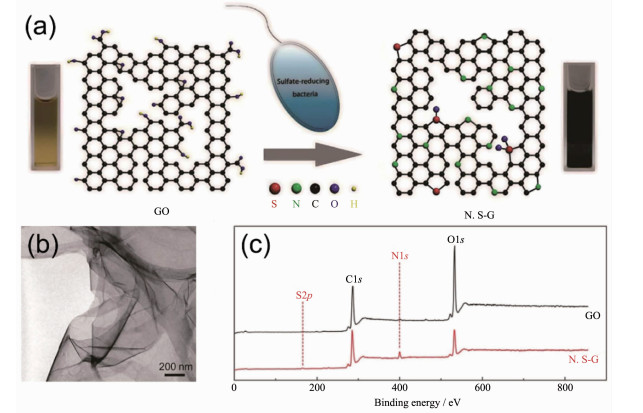
一些非金属离子也被报道可以由微生物体系合成非金属单质及其氧化物、硫化物纳米材料,特别是一些准金属元素受到了研究者们的广泛关注[15, 159]。例如,Baesman的团队[160]用2种厌氧菌合成了具有针状和球状的Te纳米颗粒;中科院合肥智能机械研究所刘锦淮的团队[161]利用B. subtilis合成出Se纳米球和纳米线并将其用于制备H2O2生物传感器;Lee的课题组[162]利用希瓦氏菌属(Shewanella sp.)在厌氧条件下合成了As2S3纳米管,随着时间的推移,纳米管的结晶度和化学成分也在不断变化,最终产生了As4S5、AsS等多种砷化物;As2O3也被报道可以由微生物合成得到[163]。基于微生物强大的生物还原能力和复杂的表面结构,近年来微生物还被用于合成碳纳米材料。Salas[164]和李轶[165]的团队利用Shewanella sp.在常温有氧条件下还原GO合成了石墨烯材料。加州大学圣克鲁兹分校李轶的团队认为Shewanella sp.在还原GO的过程中不仅外膜的细胞色素c直接介导了胞外的电子转移,而且细菌自分泌的可溶性电子介质也参与了胞外电子转移过程[165]。中科院化学研究所刘云圻院士和华中科技大学王帅的团队[110]还进一步利用硫酸盐还原菌还原GO得到了氮和硫掺杂的石墨烯材料(图 11),并将其用于制备检测Cd2+和Pb2+的电化学传感器。此外,细菌繁殖时所合成的细菌纤维素因具有复杂的三维多孔网状结构近年来被广泛用于合成三维碳纳米材料[166],微生物膜也因具有高比表面积和许多杂原子而被用于合成碳基材料。尽管微生物具有合成多种非金属纳米材料的潜力,但目前这方面的研究还较少,微生物合成其他非金属纳米材料的方法还有待开发。
图 11
4. 总结与展望
过去几十年间,人们已经利用微生物的多种生存代谢机制得到了不同种类的无机纳米材料。这些基于微生物体系合成的无机纳米材料不仅完整地保留了传统物理、化学方法合成纳米材料的光学、电学、磁学等性质,还具有环境友好、成本低廉、生物相容性好等突出优点。因此,利用微生物体系合成无机纳米材料这一领域正不断得到重视,并在生物医学、催化等方面实现了初步应用。尽管这一领域目前已经取得了巨大的进展,但未来还有许多工作需要进一步开展:
(ⅰ)深入研究微生物合成无机纳米材料的机理,精确控制无机纳米材料的形貌特征。尽管人们已经知道微生物的解毒机制和其他代谢过程在合成中起关键作用,但仍有很多微生物的合成机理尚不明确,细胞的转运机制、pH值、金属离子浓度、培养介质、材料表面的生物分子如何具体影响合成过程仍有待阐明,这导致目前微生物合成无机纳米材料的分散性、尺寸控制还不够理想。对此,已有许多团队在分子和基因水平上对微生物合成无机纳米材料的机理进行了研究。支志明[49]和Vasylevskyi[167]的团队对参与大肠杆菌和细菌G. sulfurreducens合成AgNPs的生物分子进行了精确定位,阐明了生物还原的电子传递过程,对于理解生物分子在生物还原中的作用有着重要的意义。庞代文课题组[32, 58]基于对酵母的Se还原途径和Cd解毒过程的理解,设计出了合成具有理想尺寸和发光性能的CdSe QDs的方法。Kang等[69]根据植物螯合肽的解毒机制,将植物螯合肽合成酶的基因引入大肠杆菌,其所合成的CdS纳米晶的尺寸均匀性得到了优化,证明了从基因层面调控微生物合成无机纳米材料效果的可能性。此外研究者们还一直通过基因测序等手段试图探究和模仿趋磁细菌合成高度有序磁性纳米材料的能力,但是进展仍然缓慢[10]。尽管目前人们对微生物合成机制方面的理解非常有限,但从分子和基因层面研究微生物合成机制将是合成出具有理想尺寸、形状和分散性的无机纳米材料的出路之一,故未来应当更多地在分子和基因水平上深入探索微生物合成无机纳米材料机理。通过了解生物分子的表达和作用过程,以及合成参数对生物分子的影响,我们可以在时间和空间上对微生物诱导的还原、成核、生长、稳定等过程进行选择性调控,最终像物理化学方法一样通过调节合成参数来精确控制无机纳米材料的维度、尺寸、形状和分散性等性质。
(ⅱ)提高微生物合成的效率,实现大规模生产。微生物的培养到合成过程需要花费数小时甚至数天,产品数量相对物理化学方法较少,无机纳米材料与细胞的分离过程也会增加处理难度,这些阻碍了无机纳米材料的大规模生产。未来需要深入研究稳定高效的无机纳米材料合成方法以实现大规模生产。细胞外合成具有效率高、易分离等优势[46],对此Moon等[168-170]设计了基于细胞外合成的生物反应器工艺并优化参数,实现了利用热厌氧杆菌属(Thermoanaerobacter sp.)大规模胞外合成ZnS和磁性纳米颗粒,展示了微生物合成纳米材料实现工业化应用的巨大前景。微生物生长繁殖速度的加快能显著提高无机纳米材料的产率,其中生物膜作为微生物活跃生长的聚集体,繁殖迅速,表面积大,分泌的生物分子量多,抗外界干扰能力强,是极具效率的合成载体[53],基于生物膜设计的生物反应器有望大幅缩短合成时间[51],但目前关于这方面的研究还较少。相比活细胞体系,无细胞提取物反应体系更加简单、易控制和易实现分离[46],因此利用微生物无细胞提取物进行合成也是实现工业化的有效方式。此外,Chen等[71]通过基因编辑使重组大肠杆菌中的谷胱甘肽合成酶过表达,最终CdS纳米颗粒的产量达到了对照组的2.5倍。细胞外合成、生物膜技术、无细胞提取物合成、基因编辑以及其他增强微生物发酵能力的技术能提高合成效率,在未来应当得到更多的研究,并通过取长补短充分发挥微生物合成无机纳米材料绿色、节能的优点。
(ⅲ)丰富微生物合成材料的种类。微生物对重金属的解毒过程是用于合成无机纳米材料常见的手段,这使得目前微生物合成的无机纳米材料往往集中于金属纳米材料或CdS等半导体纳米材料,对于非金属等其他无机纳米材料不易通过微生物获得,如碳纳米材料、无机盐类。迄今为止已有许多研究者采取高温处理的办法将微生物细胞转化成杂原子掺杂的碳纳米材料,或者采取生物还原由GO合成石墨烯材料,这是丰富微生物合成碳纳米材料种类的重要尝试,但合成高性能碳纳米材料方面的探索还不够。微生物还具有强大的生物矿化能力,在自然界中已广泛合成了CaCO3、Ca3(PO4)2、CaSO4等无机矿物质[171],但利用微生物合成具有复杂成分的化合物(如钙钛矿纳米材料等),以及含缺陷或掺杂的无机纳米材料的方法仍需进一步开发。基因编辑技术同样对于丰富无机纳米材料的种类具有重要意义,如Park等[70]利用能够表达其他来源的植物螯合肽合成酶和金属硫蛋白的重组大肠杆菌合成了多种新型无机纳米材料。开发新材料的合成方法同样需要对微生物合成机制有着深入理解,未来应当研究各种微生物以及相应生物分子的合成特性,充分挖掘微生物体系合成某一类无机纳米材料的能力,继而举一反三,开发出多种无机纳米材料的合成方法。
总之,利用微生物体系合成无机纳米材料具有广阔的前景。细菌、真菌等微生物因自然赋予的强大生存能力成为了无机纳米材料的合成工厂,并可能在催化、环境、医学等领域发挥重要作用。但目前微生物体系合成无机纳米材料这一领域的开发探索还不够深入,还缺少实现大规模可控合成的方法设计,在机理研究、材料性质的控制、合成效率的提高、纳米材料种类多样性等方面还有许多问题亟待解决。如何利用微生物高效地合成所需无机纳米材料是一个物理、化学、生物多学科交叉的问题,在未来需要各个学科的科学家共同努力解决。
-
-
[1]
Guru P S, Dash S. Adv. Colloid Interface Sci., 2014, 209:49-67 doi: 10.1016/j.cis.2013.12.013
-
[2]
Palmer L C, Newcomb C J, Kaltz S R, et al. Chem. Rev., 2008, 108(11):4754-4783 doi: 10.1021/cr8004422
-
[3]
Yao S S, Jin B, Liu Z M, et al. Adv. Mater., 2017, 29(14):1605903 doi: 10.1002/adma.201605903
-
[4]
Mann S. Nature, 1993, 365(6446):499-505 doi: 10.1038/365499a0
-
[5]
Addadi L, Weiner S. Angew. Chem. Int. Ed., 1992, 31(2):153-169 doi: 10.1002/anie.199201531
-
[6]
Griebler C, Lueders T. Freshwater Biology, 2009, 54(4):649-677 doi: 10.1111/j.1365-2427.2008.02013.x
-
[7]
Trofimov A A, Pawlicki A A, Borodinov N, et al. npj Comput. Mater., 2019, 5(1):4
-
[8]
Kröger N, Deutzmann R, Sumper M. Science, 1999, 286(5442):1129-1132 doi: 10.1126/science.286.5442.1129
-
[9]
Oliver S, Kuperman A, Coombs N, et al. Nature, 1995, 378(6552):47-50 doi: 10.1038/378047a0
-
[10]
Faivre D, Schüler D. Chem. Rev., 2008, 108(11):4875-4898 doi: 10.1021/cr078258w
-
[11]
Schüler D, Frankel R B. Appl. Microbiol. Biotechnol., 1999, 52(4):464-473 doi: 10.1007/s002530051547
-
[12]
Mann H, Fyfe W S. Can. J. Earth Sci., 1989, 26(12):2731-2735 doi: 10.1139/e89-234
-
[13]
Krumov N, Perner-Nochta I, Oder S, et al. Chem. Eng. Technol., 2009, 32(7):1026-1035 doi: 10.1002/ceat.200900046
-
[14]
Schultze-Lam S, Fortin D, Davis B S, et al. Chem. Geol., 1996, 132(1):171-181 doi: 10.1016/S0009-2541(96)00053-8
-
[15]
Nancharaiah Y V, Lens P N L. Trends Biotechnol., 2015, 33(6):323-330 doi: 10.1016/j.tibtech.2015.03.004
-
[16]
Li J H, Bernard S, Benzerara K, et al. Earth Planet. Sci. Lett., 2014, 400:113-122 doi: 10.1016/j.epsl.2014.05.031
-
[17]
Rahn-Lee L, Komeili A. Front. Microbiol., 2013, 4:352 doi: 10.3389/fmicb.2013.00352
-
[18]
Ray P C. Chem. Rev., 2010, 110(9):5332-5365 doi: 10.1021/cr900335q
-
[19]
Colvin V L. Nat. Biotechnol., 2003, 21(10):1166-1170 doi: 10.1038/nbt875
-
[20]
Li Y, Zhang H. Nanomedicine, 2019, 14(11):1493-1512 doi: 10.2217/nnm-2018-0346
-
[21]
Matsunaga T, Okamura Y, Tanaka T. J. Mater. Chem., 2004, 14(14):2099-2105 doi: 10.1039/b404844j
-
[22]
Darabdhara G, Das M R, Singh S P, et al. Adv. Colloid Interface Sci., 2019, 271:101991 doi: 10.1016/j.cis.2019.101991
-
[23]
Gahlawat G, Choudhury A R. RSC Adv., 2019, 9(23):12944-12967 doi: 10.1039/C8RA10483B
-
[24]
Moghaddam A B, Namvar F, Moniri M, et al. Molecules, 2015, 20(9):16540-16565 doi: 10.3390/molecules200916540
-
[25]
Beveridge T J, Murray R G. J. Bacteriol., 1980, 141(2):876-887 doi: 10.1128/JB.141.2.876-887.1980
-
[26]
Klaus T, Joerger R, Olsson E, et al. Proc. Natl. Acad. Sci. USA, 1999, 96(24):13611-13614 doi: 10.1073/pnas.96.24.13611
-
[27]
Shedbalkar U, Singh R, Wadhwani S, et al. Adv. Colloid Interface Sci., 2014, 209:40-48 doi: 10.1016/j.cis.2013.12.011
-
[28]
Poulose S, Panda T, Nair P P, et al. J. Nanosci. Nanotechnol., 2014, 14(2):2038-2049 doi: 10.1166/jnn.2014.9019
-
[29]
Huang J L, Lin L Q, Sun D H, et al. Chem. Soc. Rev., 2015, 44(17):6330-6374 doi: 10.1039/C5CS00133A
-
[30]
Dahoumane S A, Mechouet M, Wijesekera K, et al. Green Chem., 2017, 19(3):552-587 doi: 10.1039/C6GC02346K
-
[31]
Zhou J, Yang Y, Zhang C Y. Chem. Rev., 2015, 115(21):11669-11717 doi: 10.1021/acs.chemrev.5b00049
-
[32]
熊玲红, 崔然, 刘茴茴, 等.中国科学:化学, 2016, 46(2):163-172 http://www.cnki.com.cn/Article/CJFDTotal-JBXK201602003.htmXIONG Ling-Hong, CUI Ran, LIU Hui-Hui, et al. SCIENTIA SINICA Chimica, 2016, 46(2):163-172 http://www.cnki.com.cn/Article/CJFDTotal-JBXK201602003.htm
-
[33]
Reith F, Etschmann B, Grosse C, et al. Proc. Natl. Acad. Sci. USA, 2009, 106(42):17757-17762 doi: 10.1073/pnas.0904583106
-
[34]
Hulkoti N I, Taranath T C. Colloids Surf. B, 2014, 121:474-483 doi: 10.1016/j.colsurfb.2014.05.027
-
[35]
Siddiqi K S, Husen A. Nanoscale Res. Lett., 2016, 11(1):98 doi: 10.1186/s11671-016-1311-2
-
[36]
He S Y, Guo Z R, Zhang Y, et al. Mater. Lett., 2007, 61(18):3984-3987 doi: 10.1016/j.matlet.2007.01.018
-
[37]
Anil Kumar S, Abyaneh M K, Gosavi S W, et al. Biotechnol. Lett., 2007, 29(3):439-445 doi: 10.1007/s10529-006-9256-7
-
[38]
Moteshafi H, Mousavi S M, Shojaosadati S A. J. Ind. Eng. Chem., 2012, 18(6):2046-2050 doi: 10.1016/j.jiec.2012.05.025
-
[39]
Singh R, Shedbalkar U U, Wadhwani S A, et al. Appl. Microbiol. Biotechnol., 2015, 99(11):4579-4593 doi: 10.1007/s00253-015-6622-1
-
[40]
Deplanche K, Caldelari I, Mikheenko I P, et al. Microbiology, 2010, 156(9):2630-2640 doi: 10.1099/mic.0.036681-0
-
[41]
Li Y M, Li Y M, Li Q, et al. J. Nanomater., 2017, 2017:9703614
-
[42]
Narayanan K B, Sakthivel N. Adv. Colloid Interface Sci., 2010, 156(1):1-13 doi: 10.1016/j.cis.2010.02.001
-
[43]
Park T J, Lee K G, Lee S Y. Appl. Microbiol. Biotechnol., 2016, 100(2):521-534 doi: 10.1007/s00253-015-6904-7
-
[44]
Das S K, Liang J N, Schmidt M, et al. ACS Nano, 2012, 6(7):6165-6173 doi: 10.1021/nn301502s
-
[45]
Benzerara K, Miot J, Morin G, et al. C. R. Geosci., 2011, 343(2):160-167 doi: 10.1016/j.crte.2010.09.002
-
[46]
Dhillon G S, Brar S K, Kaur S, et al. Crit. Rev. Biotechnol., 2012, 32(1):49-73 doi: 10.3109/07388551.2010.550568
-
[47]
Gade A K, Bonde P, Ingle A P, et al. J. Biobased Mater. Bioenergy, 2008, 2(3):243-247 doi: 10.1166/jbmb.2008.401
-
[48]
刘紫嫣, 周豪, 沈娥, 等.微生物学通报, 2015, 42(8):1584-1592 doi: 10.13344/j.microbiol.china.140868LIU Zi-Yan, ZHOU Hao, SHEN E, et al. Microbiology China, 2015, 42(8):1584-1592 doi: 10.13344/j.microbiol.china.140868
-
[49]
Lin I W S, Lok C N, Che C M. Chem. Sci., 2014, 5(8):3144-3150 doi: 10.1039/C4SC00138A
-
[50]
He W, Zhou W J, Wang Y J, et al. Mater. Sci. Eng. C, 2009, 29(4):1348-1350
-
[51]
Kitching M, Ramani M, Marsili E. Microb. Biotechnol., 2015, 8(6):904-917 doi: 10.1111/1751-7915.12151
-
[52]
Quester K, Avalos-Borja M, Vilchis-Nestor A R, et al. PLoS One, 2013, 8(10):e77486 doi: 10.1371/journal.pone.0077486
-
[53]
Tanzil A H, Sultana S T, Saunders S R, et al. Enzyme Microb. Technol., 2016, 95:4-12 doi: 10.1016/j.enzmictec.2016.07.015
-
[54]
Bansal V, Rautaray D, Bharde A, et al. J. Mater. Chem., 2005, 15(26):2583-2589 doi: 10.1039/b503008k
-
[55]
Bansal V, Rautaray D, Ahmad A, et al. J. Mater. Chem., 2004, 14(22):3303-3305 doi: 10.1039/b407904c
-
[56]
Lee S Y, Lim J S, Harris M T. Biotechnol. Bioeng., 2012, 109(1):16-30 doi: 10.1002/bit.23328
-
[57]
Mandal D, Bolander M E, Mukhopadhyay D, et al. Appl. Microbiol. Biotechnol., 2006, 69(5):485-492 doi: 10.1007/s00253-005-0179-3
-
[58]
Cui R, Liu H H, Xie H Y, et al. Adv. Funct. Mater., 2009, 19(15):2359-2364 doi: 10.1002/adfm.200801492
-
[59]
Labrenz M, Druschel G K, Thomsen-Ebert T, et al. Science, 2000, 290(5497):1744-1747 doi: 10.1126/science.290.5497.1744
-
[60]
Kashyap P L, Kumar S, Srivastava A K, et al. World J. Microbiol. Biotechnol., 2013, 29(2):191-207 doi: 10.1007/s11274-012-1171-6
-
[61]
Das S K, Das A R, Guha A K. Small, 2010, 6(9):1012-1021 doi: 10.1002/smll.200902011
-
[62]
Srikar S K, Giri D D, Pal D B, et al. Green Sustainable Chem., 2016, 6:34-56 doi: 10.4236/gsc.2016.61004
-
[63]
Liu B, Xie J, Lee J Y, et al. J. Phys. Chem. B, 2005, 109(32):15256-15263 doi: 10.1021/jp051449n
-
[64]
Sreedharan S M, Singh S P, Singh R. Indian J. Microbiol., 2019, 59(3):321-327
-
[65]
Saxena J, Sharma M, Gupta S, et al. World J. Pharm. Pharm. Sci., 2014, 3(9):1586-1613
-
[66]
Shenton W, Douglas T, Young M, et al. Adv. Mater., 1999, 11(3):253-256 doi: 10.1002/(SICI)1521-4095(199903)11:33.0.CO;2-J
-
[67]
Bao H F, Hao N, Yang Y X, et al. Nano Res., 2010, 3(7):481-489 doi: 10.1007/s12274-010-0008-6
-
[68]
Beveridge T J. Curr. Opin. Struct. Biol., 1994, 4(2):204-212 doi: 10.1016/S0959-440X(94)90309-3
-
[69]
Kang S H, Bozhilov K N, Myung N V, et al. Angew. Chem. Int. Ed., 2008, 47(28):5186-5189 doi: 10.1002/anie.200705806
-
[70]
Park T J, Lee S Y, Heo N S, et al. Angew. Chem. Int. Ed., 2010, 49(39):7019-7024 doi: 10.1002/anie.201001524
-
[71]
Chen Y L, Tuan H Y, Tien C W, et al. Biotechnol. Progr., 2009, 25(5):1260-1266 doi: 10.1002/btpr.199
-
[72]
Mohanpuria P, Rana N K, Yadav S K. J. Nanopart. Res., 2008, 10(3):507-517 doi: 10.1007/s11051-007-9275-x
-
[73]
Gupta S K, Shukla P. Crit. Rev. Biotechnol., 2017, 37(5):672-684 doi: 10.1080/07388551.2016.1214557
-
[74]
Asmathunisha N, Kathiresan K. Colloids Surf. B, 2013, 103:283-287 doi: 10.1016/j.colsurfb.2012.10.030
-
[75]
Slocik J M, Naik R R, Stone M O, et al. J. Mater. Chem., 2005, 15(7):749-753 doi: 10.1039/b413074j
-
[76]
Huang Y, Chiang C Y, Lee S K, et al. Nano Lett., 2005, 5(7):1429-1434 doi: 10.1021/nl050795d
-
[77]
Li X Q, Xu H Z, Chen Z S, et al. J. Nanomater., 2011, 2011:270974 doi: 10.1155/2011/270974
-
[78]
Gericke M, Pinches A. Hydrometallurgy, 2006, 83(1):132-140 doi: 10.1016/j.hydromet.2006.03.019
-
[79]
Sanghi R, Verma P. Bioresour. Technol., 2009, 100(1):501-504 doi: 10.1016/j.biortech.2008.05.048
-
[80]
Shivaji S, Madhu S, Singh S. Process Biochem., 2011, 46(9):1800-1807 doi: 10.1016/j.procbio.2011.06.008
-
[81]
Kathiresan K, Manivannan S, Nabeel M A, et al. Colloids Surf. B, 2009, 71(1):133-137 doi: 10.1016/j.colsurfb.2009.01.016
-
[82]
Brown S, Sarikaya M, Johnson E. J. Mol. Biol., 2000, 299(3):725-735 doi: 10.1006/jmbi.2000.3682
-
[83]
Yong P, Rowson N A, Farr J P G, et al. J. Chem. Technol. Biotechnol., 2002, 77(5):593-601 doi: 10.1002/jctb.606
-
[84]
Ramanathan R, O'Mullane A P, Parikh R Y, et al. Langmuir, 2011, 27(2):714-719
-
[85]
Mohammed Fayaz A, Balaji K, Kalaichelvan P T, et al. Colloids Surf. B, 2009, 74(1):123-126
-
[86]
Zhang X R, He X X, Wang K M, et al. J. Nanosci. Nanotechnol., 2009, 9(10):5738-5744 doi: 10.1166/jnn.2009.1287
-
[87]
Xie J P, Lee J Y, Wang D I C, et al. J. Phys. Chem. C, 2007, 111(45):16858-16865 doi: 10.1021/jp0752668
-
[88]
Riddin T, Gericke M, Whiteley C G. Enzyme Microb. Technol., 2010, 46(6):501-505 doi: 10.1016/j.enzmictec.2010.02.006
-
[89]
He S Y, Guo Z R, Zhang Y, et al. Biotechnol. Progr., 2008, 24(2):476-480 doi: 10.1021/bp0703174
-
[90]
Zhou W J, Xiong T L, Shi C H, et al. Angew. Chem. Int. Ed., 2016, 55(29):8416-8420 doi: 10.1002/anie.201602627
-
[91]
Das S K, Das A R, Guha A K. Langmuir, 2009, 25(14):8192-8199 doi: 10.1021/la900585p
-
[92]
Lengke M F, Fleet M E, Southam G. Langmuir, 2006, 22(6):2780-2787 doi: 10.1021/la052652c
-
[93]
Durán N, Marcato P D, De Souza G I H, et al. J. Biomed. Nanotechnol., 2007, 3(2):203-208 https://www.mendeley.com/catalogue/a562b5c1-4abf-3f49-8934-1c69b9b6563b/
-
[94]
Das S K, Khan M M R, Guha A K, et al. Bioresour. Technol., 2012, 124:495-499 doi: 10.1016/j.biortech.2012.08.071
-
[95]
Vigneshwaran N, Kathe A A, Varadarajan P V, et al. Colloids Surf. B, 2006, 53(1):55-59 doi: 10.1016/j.colsurfb.2006.07.014
-
[96]
Kimber R L, Lewis E A, Parmeggiani F, et al. Small, 2018, 14(10):1703145 doi: 10.1002/smll.201703145
-
[97]
Sinha A, Khare S K. Bioresour. Technol., 2011, 102(5):4281-4284 doi: 10.1016/j.biortech.2010.12.040
-
[98]
Xiong L, Chen J J, Huang Y X, et al. Nano Energy, 2015, 12:33-42 doi: 10.1016/j.nanoen.2014.11.065
-
[99]
Liu J W, Zheng Y, Hong Z L, et al. Sci. Adv., 2016, 2(9):e1600858 doi: 10.1126/sciadv.1600858
-
[100]
Amemiya Y, Arakaki A, Staniland S S, et al. Biomaterials, 2007, 28(35):5381-5389 doi: 10.1016/j.biomaterials.2007.07.051
-
[101]
Tuo Y, Liu G F, Dong B, et al. Sci. Rep., 2015, 5:13515 doi: 10.1038/srep13515
-
[102]
Bharde A A, Parikh R Y, Baidakova M, et al. Langmuir, 2008, 24(11):5787-5794 doi: 10.1021/la704019p
-
[103]
Raliya R, Biswas P, Tarafdar J C. Biotechnol. Rep., 2015, 5:22-26 doi: 10.1016/j.btre.2014.10.009
-
[104]
Tripathi R M, Bhadwal A S, Gupta R K, et al. J. Photochem. Photobiol. B, 2014, 141:288-295 doi: 10.1016/j.jphotobiol.2014.10.001
-
[105]
Jha A K, Prasad K, Prasad K. Biotechnol. J., 2009, 4(11):1582-1585 doi: 10.1002/biot.200900144
-
[106]
Sakimoto K K, Wong A B, Yang P D. Science, 2016, 351(6268):74-77 doi: 10.1126/science.aad3317
-
[107]
Kowshik M, Deshmukh N, Vogel W, et al. Biotechnol. Bioeng., 2002, 78(5):583-588 doi: 10.1002/bit.10233
-
[108]
Ma X M, Chen H F, Yang L, et al. Angew. Chem. Int. Ed., 2011, 50(32):7414-7417 doi: 10.1002/anie.201100126
-
[109]
Sarkar J, Dey P, Saha S, et al. Micro Nano Lett., 2011, 6(8):599-602 doi: 10.1049/mnl.2011.0227
-
[110]
Guo P P, Xiao F, Liu Q, et al. Sci. Rep., 2013, 3:3499 doi: 10.1038/srep03499
-
[111]
Mukherjee P, Ahmad A, Mandal D, et al. Angew. Chem. Int. Ed., 2001, 40(19):3585-3588 doi: 10.1002/1521-3773(20011001)40:19<3585::AID-ANIE3585>3.0.CO;2-K
-
[112]
Mukherjee P, Ahmad A, Mandal D, et al. Nano Lett., 2001, 1(10):515-519 doi: 10.1021/nl0155274
-
[113]
Sundaramoorthi C, Rajasekaran A, Kalaiselvan V, et al. Int. J. Pharm. Tech. Res., 2009, 1(4):1523-1529
-
[114]
Joerger R, Klaus T, Granqvist C G. Adv. Mater., 2000, 12(6):407-409 doi: 10.1002/(SICI)1521-4095(200003)12:6<407::AID-ADMA407>3.0.CO;2-O
-
[115]
Song X J, Shi X Y. Environ. Sci. Pollut. Res., 2017, 24(3):3038-3044 doi: 10.1007/s11356-016-8076-0
-
[116]
Sen I K, Maity K, Islam S S. Carbohydr. Polym., 2013, 91(2):518-528 doi: 10.1016/j.carbpol.2012.08.058
-
[117]
Lloyd J R, Yong P, Macaskie L E. Appl. Environ. Microbiol., 1998, 64(11):4607-4609 doi: 10.1128/AEM.64.11.4607-4609.1998
-
[118]
Lengke M F, Fleet M E. Southam G. Langmuir, 2006, 22(17):7318-7323 doi: 10.1021/la060873s
-
[119]
Søbjerg L S, Lindhardt A T, Skrydstrup T, et al. Colloids Surf. B, 2011, 85(2):373-378 doi: 10.1016/j.colsurfb.2011.03.014
-
[120]
Puja P, Kumar P. Spectrochim. Acta Part A, 2019, 211:94-99 doi: 10.1016/j.saa.2018.11.047
-
[121]
Xiong L, Zhang X, Huang Y X, et al. ACS Appl. Nano Mater., 2018, 1(4):1467-1475 doi: 10.1021/acsanm.7b00322
-
[122]
Song X J, Shi X Y, Yang M. IET Nanobiotechnol., 2017, 12(4):441-445
-
[123]
Prasad K, Jha A K, Kulkarni A R. Nanoscale Res. Lett., 2007, 2(5):248-250 doi: 10.1007/s11671-007-9060-x
-
[124]
Mullen M D, Wolf D C, Ferris F G, et al. Appl. Environ. Microbiol., 1989, 55(12):3143-3149 doi: 10.1128/AEM.55.12.3143-3149.1989
-
[125]
Godipurge S S, Yallappa S, Biradar N J, et al. Enzyme Microb. Technol., 2016, 95:174-184 doi: 10.1016/j.enzmictec.2016.08.006
-
[126]
Nair B, Pradeep T. Cryst. Growth Des., 2002, 2(4):293-298
-
[127]
Senapati S, Ahmad A, Khan M I, et al. Small, 2005, 1(5):517-520 doi: 10.1002/smll.200400053
-
[128]
Zheng D Y, Hu C G, Gan T, et al. Sens. Actuators B, 2010, 148(1):247-252 doi: 10.1016/j.snb.2010.04.031
-
[129]
Han R S, Song X, Wang Q H, et al. J. Chem. Technol. Biotechnol., 2019, 94(10):3375-3383 doi: 10.1002/jctb.6150
-
[130]
Blakemore R. Science, 1975, 190(4212):377-379 doi: 10.1126/science.170679
-
[131]
Kumar U, Shete A, Harle A S, et al. Chem. Mater., 2008, 20(4):1484-1491 doi: 10.1021/cm702727x
-
[132]
Bharde A, Rautaray D, Bansal V, et al. Small, 2006, 2(1):135-141 doi: 10.1002/smll.200500180
-
[133]
Byrne J M, Coker V S, Cespedes E, et al. Adv. Funct. Mater., 2014, 24(17):2518-2529 doi: 10.1002/adfm.201303230
-
[134]
Wu L H, Mendoza-Garcia A, Li Q, et al. Chem. Rev., 2016, 116(18):10473-10512 doi: 10.1021/acs.chemrev.5b00687
-
[135]
Coker V S, Bennett J A, Telling N D, et al. ACS Nano, 2010, 4(5):2577-2584 doi: 10.1021/nn9017944
-
[136]
Watson J H P, Ellwood D C, Soper A K, et al. J. Magn. Magn. Mater., 1999, 203(1):69-72 doi: 10.1016/s0304-8853(99)00191-2
-
[137]
Watson J H P, Ellwood D C. Miner. Eng., 1994, 7(8):1017-1028 doi: 10.1016/0892-6875(94)90030-2
-
[138]
Lisy M R, Hartung A, Lang C, et al. Invest. Radiol., 2007, 42(4):235-241 doi: 10.1097/01.rli.0000255832.44443.e7
-
[139]
Suzuki Y, Kelly S D, Kemner K M, et al. Nature, 2002, 419(6903):134 doi: 10.1038/419134a
-
[140]
Karunagaran V, Rajendran K, Sen S. Int. J. Nanosci., 2017, 16(05n06):1750018 doi: 10.1142/S0219581X17500181
-
[141]
Atla S B, Chen Y J, Chiu H W, et al. Mater. Lett., 2016, 167:238-241 doi: 10.1016/j.matlet.2015.12.123
-
[142]
Uddin I, Adyanthaya S, Syed A, et al. J. Nanosci. Nanotechnol., 2008, 8(8):3909-3913 doi: 10.1166/jnn.2008.179
-
[143]
Cuevas R, Durán N, Diez M, et al. J. Nanomater., 2015, 16(1):57 doi: 10.1155/2015/789089
-
[144]
Dameron C T, Reese R N, Mehra R K, et al. Nature, 1989, 338(6216):596-597 doi: 10.1038/338596a0
-
[145]
Cunningham D P, Lundie L L. Appl. Environ. Microbiol., 1993, 59(1):7-14 doi: 10.1016/S0065-2164(08)70598-7
-
[146]
Kowshik M, Vogel W, Urban J, et al. Adv. Mater., 2002, 14(11):815-818 doi: 10.1002/1521-4095(20020605)14:11<815::AID-ADMA815>3.0.CO;2-K
-
[147]
Mala J G S, Rose C. J. Biotechnol., 2014, 170:73-78 doi: 10.1016/j.jbiotec.2013.11.017
-
[148]
Cuéllar-Cruz M, Lucio-Hernández D, Martínez-ángeles I, et al. Microb. Biotechnol., 2017, 10(2):405-424
-
[149]
Bai H J, Zhang Z M. Mater. Lett., 2009, 63(9):764-766 doi: 10.1016/j.matlet.2008.12.050
-
[150]
Debabov V G, Voeikova T A, Shebanova A S, et al. Nanotechnol. Russ., 2013, 8(3):269-276 doi: 10.1134/S1995078013020043
-
[151]
Sathyavathi S, Manjula A, Rajendhran J, et al. Indian J. Exp. Biol., 2013, 51:973-978 doi: 10.1038/gt.2013.37
-
[152]
Yue L, Wu Y, Liu X, et al. Biotechnol. Progr., 2014, 30(4):960-966
-
[153]
Hosseini M R, Schaffie M, Pazouki M, et al. Mater. Sci. Semicond. Process., 2013, 16(2):250-255 doi: 10.1016/j.mssp.2012.11.002
-
[154]
Su Y L, Du Q Q, Qu X C, et al. RSC Adv., 2016, 6(34):28187-28193 doi: 10.1039/C5RA26714E
-
[155]
Wang L Q, Pang Q, Song Q Q, et al. Fuel, 2015, 140:267-274 doi: 10.1016/j.fuel.2014.09.107
-
[156]
Wang B, Liu P, Jiang W G, et al. Angew. Chem. Int. Ed., 2008, 47(19):3560-3564 doi: 10.1002/anie.200704718
-
[157]
Rautaray D, Sanyal A, Adyanthaya S D, et al. Langmuir, 2004, 20(16):6827-6833 doi: 10.1021/la049244d
-
[158]
Bansal V, Poddar P, Ahmad A, et al. J. Am. Chem. Soc., 2006, 128(36):11958-11963 doi: 10.1021/ja063011m
-
[159]
Woolfolk C A, Whiteley H R. J. Bacteriol., 1962, 84(4):647-658 doi: 10.1128/JB.84.4.647-658.1962
-
[160]
Baesman S M, Bullen T D, Dewald J, et al. Appl. Environ. Microbiol., 2007, 73(7):2135-2143 doi: 10.1128/AEM.02558-06
-
[161]
Wang T T, Yang L B, Zhang B C, et al. Colloids Surf. B, 2010, 80(1):94-102
-
[162]
Lee J H, Kim M G, Yoo B, et al. Proc. Natl. Acad. Sci. USA, 2007, 104(51):20410-20415 doi: 10.1073/pnas.0707595104
-
[163]
Huang J H. Water Air Soil Pollut., 2014, 225(2):1848 doi: 10.1007/s11270-013-1848-y
-
[164]
Salas E C, Sun Z Z, Lüttge A, et al. ACS Nano, 2010, 4(8):4852-4856 doi: 10.1021/nn101081t
-
[165]
Wang G M, Qian F, Saltikov C W, et al. Nano Res., 2011, 4(6):563-570 doi: 10.1007/s12274-011-0112-2
-
[166]
Wu Z Y, Liang H W, Chen L F, et al. Acc. Chem. Res., 2016, 49(1):96-105 doi: 10.1021/acs.accounts.5b00380
-
[167]
Vasylevskyi S I, Kracht S, Corcosa P, et al. Angew. Chem. Int. Ed., 2017, 56(21):5926-5930 doi: 10.1002/anie.201702621
-
[168]
Moon J W, Ivanov I N, Joshi P C, et al. Acta Biomater., 2014, 10(10):4474-4483 doi: 10.1016/j.actbio.2014.06.005
-
[169]
Moon J W, Phelps T J, Fitzgerald Jr C L, et al. Appl. Microbiol. Biotechnol., 2016, 100(18):7921-7931 doi: 10.1007/s00253-016-7556-y
-
[170]
Moon J W, Rawn C J, Rondinone A J, et al. J. Ind. Microbiol. Biotechnol., 2010, 37(10):1023-1031 doi: 10.1007/s10295-010-0749-y
-
[171]
Bansal V, Bharde A, Ramanathan R, et al. Adv. Colloid Interface Sci., 2012, 179:150-168 doi: 10.1016/j.cis.2012.06.013
-
[1]
-

图 2 无机纳米材料的生物还原合成示意图
Figure 2 Biosynthesis of inorganic nanomaterials mediated by bioreduction
Biosynthesis of inorganic nanomaterials mediated by bioreduction can be classified into extracellular synthesis and intracellular synthesis; Metal ions are reduced followed by nucleation and growth of nanoparticles; Dispersed nanoparticles are obtained after stabilization by biomolecules
图 3 (a) 真菌R. oryzae合成AuNPs的机理图[44]; (b)大肠杆菌细胞色素c亚基NapC介导的AgNPs合成机理图[49]
Figure 3 (a) Proposed mechanism of AuNPs biosynthesis by fungi R. oryzae[44]; (b) Proposed mechanism of AgNPs biosynthesis mediated by c-type cytochrome subunit NapC in E. coli strain[49]
MOX: oxidized Au3+ ion; MRED: reduced metallic Au; Red: reduced protein or enzyme; Ox: oxidized protein or enzyme
图 6 (a) Au@NC的合成过程示意图[90]; (b) Au@NC的SEM图中的纤维形态的Au@NC及亮点状的AuNPs[90]; (c) Au@NC的TEM图中的AuNPs负载于NC上且粒径为10~40 nm[90]
Figure 6 (a) Preparation of N-doped carbon-supported AuNPs (Au@NC)[90]; (b)Fibrous morphology of Au@NC and bright Au dots in SEM image[90]; (c) AuNPs with sizes ranging from 10 to 40 nm loaded on N-doped carbon (NC) in TEM image[90]
AuNPs were synthesized on the cell surface and in the cell, then Au/microbe composite was further calcined to prepare Au@NC as an electrocatalyst for hydrogen evolution reaction andn oxygen evolution reaction
图 7 (a) 由S. oneidensis合成负载Pd纳米颗粒的杂原子掺杂介孔碳材料示意图[121]; (b) 400 ℃高温活化后S. oneidensis细胞和Pd纳米颗粒的SEM图[121]; (c) 400 ℃高温活化后S. oneidensis细胞和Pd纳米颗粒的TEM图[121]; (d~f) S. oneidensis在细胞内大量合成的CuNPs的TEM图[96]
Figure 7 (a) Preparation of heteroatom-doped porous carbon materials with Pd nanoparticles synthesized by S. oneidensis[121]; (b) SEM image of Pd nanoparticles and S. oneidensis cells after activation at 400 ℃[121]; (c) TEM image of Pd nanoparticles and S. oneidensis cells after activation at 400 ℃[121]; (d~f) TEM images of large amounts of CuNPs synthesized in S. oneidensis cytoplasm[96]
图 10 (a) 酵母合成CdSe QDs的机理图[58]; (b~d)经Cd2+孵育(b) 12、(c) 24和(d) 40 h后酵母的共聚焦显微镜荧光图像及其相应的原位荧光光谱[58]
Figure 10 (a) Proposed mechanism of CdSe QDs biosynthesis by yeast[58]; (b~d) In situ laser confocal scanning microscopy photographs and corresponding fluorescence spectra of yeasts after incubation with Cd2+ for (b) 12, (c) 24, and (d) 40 h, respectively[58]
GSH: glutathione; Diameters(D) of CdSe QDs increased gradually after incubation with Cd2+ for a period of time; In situ laser confocal scanning microscopy photographs and corresponding fluorescence spectra show that yeasts emitted green fluorescence, yellow fluorescence and red fluorescence after incubation with Cd2+ for 12, 24 and 40 h respectively

图 11 (a) 硫酸盐还原菌通过生物还原合成氮和硫掺杂的石墨烯材料(N, S-G)的示意图[110]; (b) N, S-G的TEM图[110]; (c) GO和N, S-G的XPS图谱[110]
Figure 11 (a) Preparation of nitrogen and sulfur-doped graphene (N, S-G) by microbial reduction of sulfate-reducing bacteria[110]; (b) TEM image of N, S-G[110]; (c) XPS survey spectra of GO and N, S-G[110]
表 1 一些微生物合成的无机纳米材料及其性质
Table 1. Some inorganic nanomaterials synthesized by microbes and their properties
Microorganism Product Size / nm Shape Application Ref. Pycnoporus sanguineus Au 10~40 Spherical Electrocatalyst [90] Rhizopus oryzae Au 10 Spherical Water purification [91] Rhizopus oryzae Au Diverse sizes Diverse shapes — [61] Plectonema boryanum Au 25 Cubic — [92] Fusarium oxysporum Ag 1.6 Spherical Antibacterial [93] Rhizopus oryzae Ag 15 Spherical Water purification [94] Phaenerochaete chrysosporium Ag 50~200 Pyramidal — [95] Pseudomonas stutzeri Ag 200 Diverse shapes — [26] Shewanella oneidensis Cu 20~50 Spherical Catalyst [96] Enterobacter sp. Hg 2~5 Spherical — [97] Shewanella sp. Pd 13 — Electrocatalyst [98] Sulfate-reducing bacteria Pt 50~300 Rectangular — [88] Shewanella oneidensis Pd-Au alloy 4.95 — Electrocatalyst [99] Magnetospirillum magneticum Fe3O4 20~30 Cubic — [100] Shewanella oneidensis Pd/Fe3O4, Au/Fe3O4, PdAu/Fe3O4 3~15 — Catalyst [101] Actinobacter sp. γ-Fe2O3, Fe3S4 5~7 — — [102] Fusarium oxysporum TiO2, SiO2 5~15 Irregular or spherical — [54] Aspergillus flavus TiO2 12~15 — Photosynthesis [103] Bacillus licheniformis ZnO 200~1000 Flower Photocatalyst [104] Lactic acid Bacillus Sb2O3 3~12 Cubic — [105] Moorella thermoacetica CdS < 10 — Photosynthesis [106] Schizosaccharomyces pombe CdS 2~2.5 Hexagonal Diode [107] Sulfate-reducing bacteria ZnS 2~5 Spherical — [59] Yeast CdTe 2~3.6 — Bio-imaging [67] Recombinant Escherichia coli CdSeZn, PrGd, CdCs, FeCo etc. Diverse sizes Atypical shapes — [70] Yeast CaCO3 4~6 — Drug delivery [108] Bacillus subtilis Se 50~400 Spherical Biosensor [109] Sulfate-reducing bacteria Graphene — — Electrocatalyst [110] —: Not reported  下载: 导出CSV
下载: 导出CSV
-













 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 165
- 文章访问数: 9432
- HTML全文浏览量: 1687
