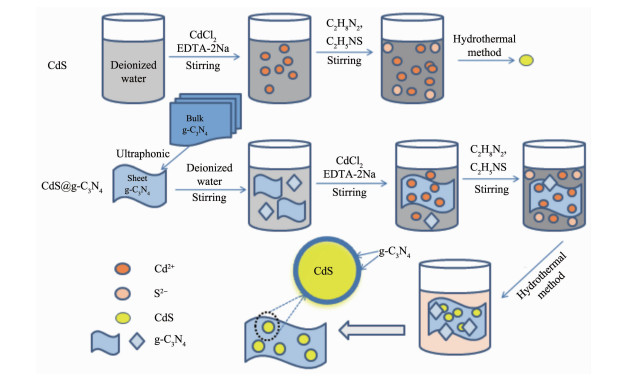
图 1.
CdS@g-C3N4复合核壳纳米微粒的制备示意图
Figure 1.
Graphical illustration for the synthesis of CdS@g-C3N4 core-shell structure composite

近年来,具有可见光响应的复合核壳结构光催化材料因其特殊的反应空间及核壳间的协同作用,受到了越来越多的关注[1-2]。核壳结构材料由中心粒子和包覆层构成,因外壳材料对内核中心材料的保护,有效避免中心粒子团聚、失活现象,比中心粒子具有更高的比表面积和光稳定性[3]。目前,人们已开发了多种制备纳米复合核壳结构材料的方法。苏进展等[2]采用超声喷雾热分解-水浴两步法制备了核壳结构CdS/ZnS微米球光催化剂。Liu等[4]采用化学吸附法制备了新型CdS@g-C3N4核壳结构光催化剂,并证实了这些复合核壳结构材料的光催化性能明显高于单组分材料。
CdS是禁带宽度为2.42 eV的直接带隙半导体[5],g-C3N4是禁带宽度为2.69 eV的间接半导体[6],2种材料都具有良好的半导体性能和可见光活性,被认为是很有前景的光催化材料,在降解水中污染物和光解水制氢方面备受青睐[7-8]。但CdS易发生光腐蚀,g-C3N4光利用率较低,两者光生电子-空穴对都易复合,导致光催化效率降低[9-10]。用能带结构匹配的2个不同组分的半导体进行耦合形成异质结构,有利于激子在异质界面聚集并快速分离,是提高两者光催化效率和稳定性的有效策略[11-12],Wang等[13]分别用三聚氰胺和尿素作为单体通过有机交联反应设计合成了基于石墨烯型的g-C3N4的CN-T/CN-M同型异质结构,有效促进了激子分离和电荷转移,提高了g-C3N4的产氢效率和稳定性。Wang等[14]还分别用溶剂热法和浸渍结合光沉积法把CdS纳米粒子负载在具有三嗪结构的有机骨架(CTF-1)上,构筑得到了CdS NPs/CTF-1异型异质结构,其具有比纯CdS、CTF-1更高的水产氢率和稳定性。CdS和g-C3N4具有匹配的能带结构,构筑CdS/g-C3N4复合核壳结构,可促进激子在CdS和g-C3N4界面转移,抑制S2-的自氧化致CdS发生光腐蚀,从而克服CdS、g-C3N4各自的缺点。Yin等[15]采用超声-化学吸附两步法合成了一系列空心CdS@g-C3N4核壳球纳米复合材料,空心CdS@g-C3N4核壳结构能显著提高CdS的光催化产氢率和光稳定性。Li等[16]采用水热法制备了CdS@g-C3N4核壳纳米线阵列结构,CdS@g-C3N4的光催化产氢率和光稳定性明显高于单一CdS。
我们采用简单水热法,在乙二胺、乙二胺四乙酸二钠盐(EDTA-2Na)的作用下,以硫代乙酰胺、氯化镉和g-C3N4纳米片为反应物,成功构筑了以超薄g-C3N4纳米片层为壳、以CdS纳米粒为核的CdS@g-C3N4核壳纳米复合物,并在可见光照下考察了其光催化降解罗丹明B (RhB)活性和重复使用性。
图 1为CdS@g-C3N4的合成示意图。晶体在不同条件下表现出的结晶形态不仅与晶体内部结构有关,而且受外部生长环境的影响,CdS结晶形态主要由正负极面和柱面的暴露程度所决定[17]。因CdS是极性晶体,其生长基元的中心Cd原子分布不对称,使CdS晶体的正负极面上都易叠合,诱导CdS晶体沿着c轴方向生长。当反应体系仅存在EDTA-2Na、乙二胺时,乙二胺、EDTA-2Na先与Cd2+分别形成Cd(en)32+、Cd(EDTA)62+等配离子,再与硫代乙酰胺释放出的S2-发生反应[18]。配离子产生的空间位阻效应抑制了CdS的形核生长,最终生成聚集态的CdS纳米粒子。当反应体系同时存在EDTA-2Na、乙二胺和g-C3N4时,带正电荷的Cd(en)32+、Cd(EDTA)62+等配离子先与含有大π键结构的g-C3N4产生共轭效应,使其极易吸附于g-C3N4基层,然后与硫代乙酰胺释放出的S2-发生反应。因空间位阻效应抑制了CdS的形核生长,促进了许多CdS小晶粒的生成;同时,因氨基化而带正电荷的球状CdS纳米粒与带负电荷的片状g-C3N4薄层通过简单静电相互作用,驱使以CdS为内核g-C3N4为外壳的一个自组装过程,最终形成CdS@g-C3N4复合核壳结构[19]。
三聚氰胺(C3H6N6,CP)、氯化镉(CdCl2,AR)均购自山东西亚化学工业有限公司;EDTA-2Na (C10H14N2O8Na2·2H2O,AR),购自广东光华科技股份有限公司;硫代乙酰胺(C2H5NS,AR)、乙二胺(C2H8N2,AR)、异丙醇((CH3)2CHOH,AR)、无水乙醇(C2H5OH,AR),均购自西陇科学股份有限公司;实验用水为去离子水。
采用热聚合法制备g-C3N4并用超声处理。称取3 g干燥的三聚氰胺于瓷坩埚中,加盖,放入马弗炉中于550 ℃下煅烧4 h,冷却、研磨得到淡黄色g-C3N4粉末,放入装有40 mL异丙醇的烧杯中,超声6 h,离心,收集沉淀,干燥,所得g-C3N4待用。
取0.15 g g-C3N4溶于装有39 mL去离子水的烧杯中,加入2 mmol CdCl2、0.2 g EDTA-2Na(0.53 mmol)并搅拌;再加入2 mmol硫代乙酰胺、1 mL乙二胺,搅拌。将上述混合液转入60 mL聚四氟乙烯反应釜内衬,180 ℃下水热反应4 h。冷却,抽滤,产物用去离子水和无水乙醇交替清洗3次,干燥,得CdS@g-C3N4样品。
上述体系中不加g-C3N4,可得纯的CdS粉体。
采用德国Bruker D8 Advance 25型X射线衍射仪(XRD,Cu靶Kα射线为激发源,波长λ=0.154 056 nm,管电压40 kV,管电流40 mA,步长0.02°,扫描范围2θ=10°~70°,扫描速度0.1°·min-1)测样品物相结构。美国Frontier型傅里叶变换傅里叶红外光谱仪(FTIR)测样品的官能团,日本JSM-6700F型扫描电子显微镜(SEM,加速电压10 kV)和日本Hitachi-7650型透射电子显微镜(TEM,加速电压120 kV)测样品微观形貌,ESCALab 250型能谱仪(EDS)测样品成分,美国ASAP 2460型比表面积与孔隙度分析仪测样品的比表面积(测定前,样品于300 ℃下真空脱气处理6 h),采用Elementar Analysensysteme有限公司的vario EL cube进行元素分析,X射线光电子能谱(XPS)使用赛默飞世尔科技有限公司的Thermo ESCALAB250进行测定,系统射线源为Cu Kα射线,结合能参照C1s标准峰(284.6 eV)以降低样品的电荷效应,采用CasaXPS进行曲线拟合,相对灵敏度因子和非对称性函数取自PHIESC手册。Maya Pro2000型紫外-可见漫反射光谱仪(UV-Vis-DRS,扫描范围300~700 nm,以BaSO4为标准白板)和RF-5301PC型荧光光谱分析仪(激发波长为340 nm)测样品光谱特性。光电性能用上海CHI660E电化学工作站测试(标准三电极系统,其中,甘汞电极(饱和KCl溶液)为参比电极,铂丝为对电极,制备的样品电极为工作电极,0.5 mol·L-1的Na2SO4作为电解液,氙灯为光源。工作电极的制作方法如下:1 cm2 ITO玻璃上先涂一层导电银膏,再均匀涂布样品,后放入真空干燥箱60 ℃干燥过夜。)。
以RhB的光降解评价所制备样品的光催化活性。光催化反应在光化学反应仪(TL-GHX-V型,江苏天翎仪器有限公司)中进行,将50 mL初始浓度为10 mg·L-1的RhB溶液倒入石英管中,再往其中加入50 mg催化剂样品,先暗箱反应30 min,达吸附平衡后,开启氙灯(PLS-LAX300ADJ型,江苏天翎仪器有限公司,1 000 W),功率调至300 W,光照2 h,每隔0.5 h用滴管取5 mL反应液置离心管内,离心,取上层清液,用紫外-可见分光光度计(UV-2000型,上海精密科学仪器有限公司)测其在554 nm处的吸光度,通过RhB标准曲线求出对应的浓度。采用如下公式计算降解率:η=(C0-Ct)/C0×100%(η为降解率,C0为初始RhB浓度,Ct为光照t时后的RhB浓度)。
收集光催化反应后的催化剂,用去离子水洗涤数次,烘干后参照上述步骤重复光催化过程,循环使用3次。
图 2是g-C3N4、CdS及CdS@g-C3N4的XRD图。可以看出,g-C3N4在13.1°和27.7°处分别出现一个较弱和较强的特征峰,分别对应于(PDF No.87-1526)中(100)和(002)晶面,其中27.7°处峰由g-C3N4共轭芳香族堆垛形成[8]。CdS在2θ为24.8°、26.6°、28.2°、36.7°、43.8°、48.1°和52.0°处的衍射峰归属于六方相CdS(PDF No.77-2306),分别对应其(100)、(002)、(101)、(102)、(110)、(103)、(112)晶面[5]。CdS@g-C3N4复合材料的XRD图与CdS的相似,未出现其它杂峰。因复合材料中g-C3N4含量较低,g-C3N4的特征衍射峰不太明显。XRD局部放大图显示(图 2b),CdS@g-C3N4中CdS的衍射峰有较为明显的宽化,且向大角度出现了一定程度的偏移,可能是因为CdS@g-C3N4中CdS与g-C3N4存在相互作用,抑制了CdS形核生长,CdS颗粒尺寸变小所致[14, 20-21]。
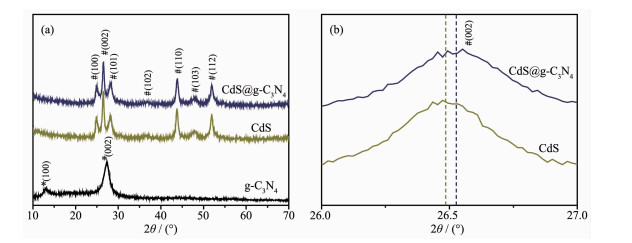
图 3分别给出了g-C3N4、CdS、CdS@g-C3N4的SEM图。由图 3可知,g-C3N4为大小不一的聚集态片状结构,表面光滑;纯CdS为圆球形颗粒,呈聚集状,粒径在20~30 nm之间;CdS@g-C3N4中CdS粒径较纯CdS小,分散性也较纯CdS更好。原因可能是,当体系中加入乙二胺、EDTA和g-C3N4后,乙二胺、EDTA与Cd2+形成的Cd(en)32+、Cd(EDTA)62+等配离子能与g-C3N4发生相互作用,抑制CdS的成核生长,导致CdS粒径变小,同时生成的CdS可与g-C3N4通过静电相互作用有效抑制CdS颗粒团聚。

图 4为CdS@g-C3N4的TEM和HRTEM图。从TEM图可看出,CdS@g-C3N4中CdS是粒径为15~25 nm的纳米颗粒,均匀分布在片状g-C3N4层中。从HRTEM图可看出,g-C3N4和CdS具有取向不同的晶格条纹,存在明显的异质界面,形成了核壳结构[22-24],内核CdS的晶面间距为0.353 nm,对应着CdS的(101)晶面[25],g-C3N4的晶面间距为0.323 nm,对应g-C3N4的(002)晶面[26],包覆在CdS外层。
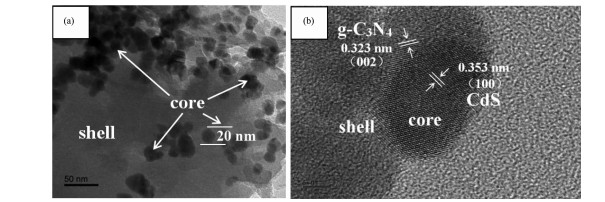
图 5是CdS@g-C3N4复合核壳结构的EDS面扫描图。图中包含均匀分布的Cd、S、C、N四种元素,且Cd与S分布相似,C与N分布相似,说明CdS@g-C3N4是以CdS和g-C3N4的形式共存[27]。
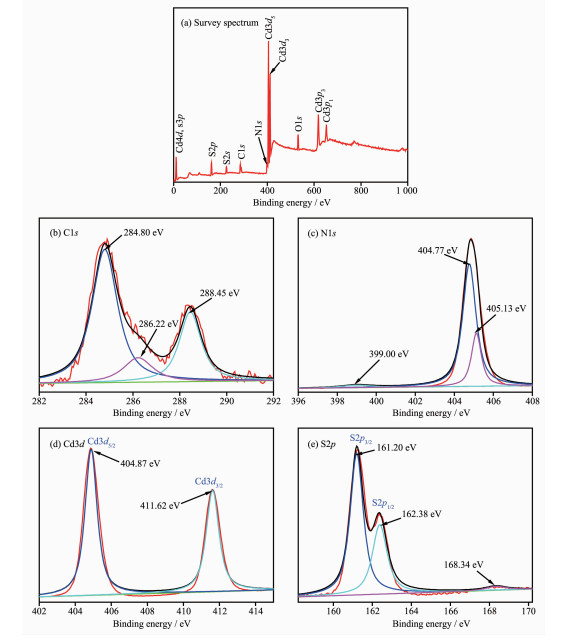
图 6(a)是CdS@g-C3N4样品的XPS全谱图,该图表明CdS@g-C3N4复合核壳结构主要由C、N、Cd、S及少量O组成,O的存在可能是制备过程中吸附在催化剂表面的H2O或-OH所致[28]。图 6(b)是C1s谱,在284.80 eV处峰归属于sp2 C-C键,286.22 eV处峰归属于CdS@g-C3N4表面缺陷的sp3配位C键,288.45 eV归属于芳香环中的sp2 N-C=N键[29]。图 6(c)是N1s谱,位于399.00 eV处弱峰归属于三嗪环(C-N=C)中的sp2 N键,404.77 eV处强峰归属于N-(C)3基团,405.13 eV处的弱峰则表明存在氨基(C-N-H)。图 6(d)是Cd3d谱,在404.87、411.62 eV处峰分别归属于Cd3d5/2、Cd3d3/2,说明CdS@g-C3N4中Cd元素主要以Cd2+的形式存在。图 6(e)的S2p谱中位于161.20、162.38 eV处峰分别归属于CdS纳米粒子中的S2p3/2、S2p1/2,说明S元素主要以S2-的形式存在;位于168.34 eV处的弱峰归属于S-C键,这应是CdS与g-C3N4存在共轭效应所引起[4, 30]。
图 7是g-C3N4、CdS、CdS@g-C3N4的FT-IR谱图。对于纯CdS,位于619 cm-1处的吸收峰属于Cd-S拉伸振动。纯g-C3N4的中,在807 cm-1处的吸收峰为s-三嗪结构的对称伸缩振动,887 cm-1处峰为NH弯曲振动[31]。强吸收带区域(1 200~1 650 cm-1)在1 242、1 326、1 413、1 572和1 642 cm-1处有特征峰,可归属于C-N杂环的典型拉伸振动,2 900~ 3 200 cm-1的宽吸收带属于N-H和O-H拉伸,说明热聚合法制备的g-C3N4残留部分-NH2,且在表面吸附了少量水分子[29]。纯CdS和纯g-C3N4中的主要特征峰同时出现在CdS@g-C3N4谱图中,说明CdS和g-C3N4共存于CdS@g-C3N4复合材料中[32]。CdS@g-C3N4相对于g-C3N4在807 cm-1处的吸收峰产生了微弱红移,并且C-N杂环振动峰弱于纯的g-C3N4,这表明CdS和g-C3N4之间可能存在相互作用,从而削弱了C和N原子之间的作用力[14]。
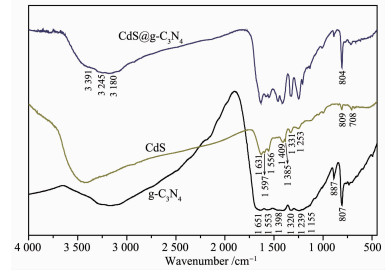
图 8是CdS、g-C3N4和CdS@g-C3N4的N2吸附-脱附等温线。由图可知,CdS、g-C3N4与CdS@g-C3N4属于Ⅳ型等温线,CdS的N2吸附-脱附曲线不存在明显回滞环,而g-C3N4与CdS@g-C3N4的N2吸附-脱附曲线中均能看到H3型回滞环,由此可以看出CdS@g-C3N4具有核壳结构交错堆积形成的空间介孔。CdS、g-C3N4比表面积分别为4、9 m2·g-1,而具有核壳结构的CdS@g-C3N4比表面积达56 m2·g-1,是纯CdS的14.0倍,这可能是CdS和g-C3N4存在的相互作用,使CdS纳米微粒对g-C3N4片层起到“剥离”作用,增大了g-C3N4层间距[33-34];还可能是乙二胺、EDTA-2Na、g-C3N4所产生的空间位阻效应抑制了CdS晶粒的生长,促进了许多CdS小晶粒的生成[35-36]。
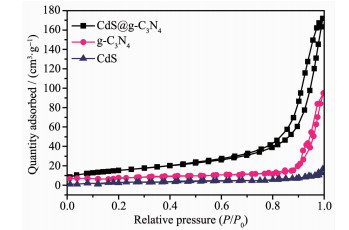
图 9(a)为g-C3N4、CdS和CdS@g-C3N4的UV-Vis DRS谱图。由图可知,纯CdS在紫外光区和可见光区都有较强吸收,吸收带边约在517 nm,纯g-C3N4的吸收带边约在420 nm。CdS@g-C3N4复合材料的吸收曲线与CdS很相似,吸收强度略低于CdS,吸收带边约在513 nm,发生了蓝移现象。这可能是因为g-C3N4与CdS复合,二者之间产生共轭效应,g-C3N4提供了大量的CN等生色基团,使得CdS@g-C3N4复合材料的吸收带边相对CdS蓝移[37]。
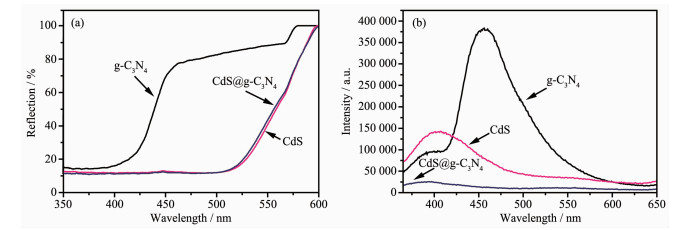
图 9(b)为g-C3N4、CdS和CdS@g-C3N4的光致发光(PL)光谱图。纯CdS在392 nm附近有较强的荧光发射峰,纯g-C3N4在470 nm附近有很强的荧光发射峰。而CdS@g-C3N4复合材料在392 nm附近的荧光发射峰强度远弱于CdS、g-C3N4,发生明显的猝灭现象[38]。这可能是g-C3N4与CdS存在相互作用,二者形成的异质结构为载流子迁移提供一个平台,使二者界面上的电荷存在高效地分离和转移。因此,CdS@g-C3N4可有效抑制光生电子-空穴对的复合[39],使发光强度显著降低。
图 10(a)是g-C3N4、CdS和CdS@g-C3N4在可见光照射下的电流-时间(I-t)曲线。3个样品都有明显的光电流响应,且CdS@g-C3N4复合核壳结构表现出最大的光电流强度,表明光生电子-空穴对复合率小,电荷分离效率高[40]。
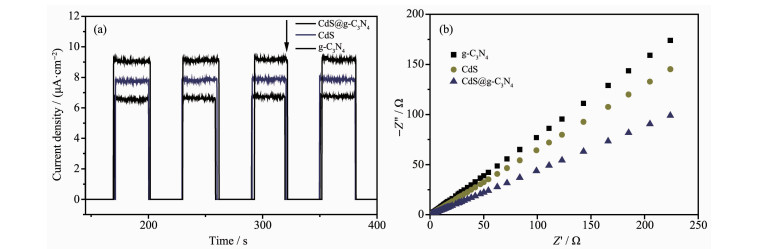
图 10(b)是g-C3N4、CdS和CdS@g-C3N4在可见光照射下的电化学阻抗谱(EIS)曲线,采用Z-view软件进行拟合。由图可知,CdS@g-C3N4的EIS阻抗谱的电荷转移电阻(Rt)数值明显低于g-C3N4、CdS,表明CdS@g-C3N4电极上电荷传输速率最快[41]。CdS@g-C3N4复合核壳结构提高了光生电子-空穴对的分离速率,增强了光催化活性。
图 11(a)为不同条件下RhB的光催化降解情况对比。当可见光照2 h,不加光催化剂时,RhB几乎不降解,说明RhB在可见光下稳定性很好。以CdS、g-C3N4和CdS@g-C3N4为光催化剂时,RhB的降解率分别为72.0%、48.4%和95.2%,说明CdS@g-C3N4复合核壳结构的光催化性能明显高于纯CdS或g-C3N4。原因是CdS@g-C3N4复合核壳结构的比表面积远大于纯CdS或g-C3N4,这增加了反应活性位点,促进了反应物的吸附及产物的扩散[42]。此外,CdS@g-C3N4复合核壳结构的形成,促进了光生载流子分离和迁移,有利于可见光催化活性的增强[29]。
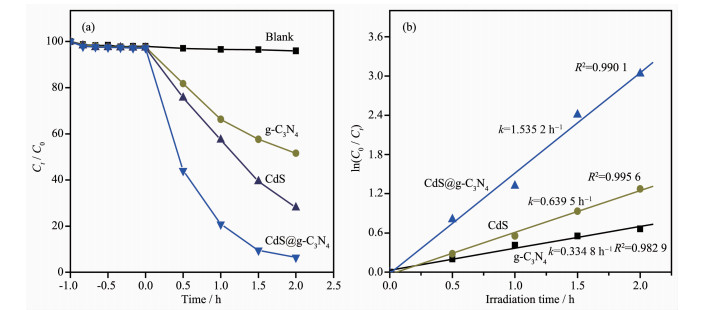
图 11(b)为RhB水溶液光催化降解反应动力学分析。由图可知,在反应达到吸附平衡后,以CdS、g-C3N4和CdS@g-C3N4复合核壳结构为光催化剂时,RhB降解反应的ln(C0/Ct)与t呈线性关系,回归系数均大于0.982 9,说明该反应较好地符合一级动力学模型[43]。CdS@g-C3N4复合核壳结构作为催化剂时,RhB表观降解速率常数k为1.535 2 h-1,分别是纯g-C3N4、CdS为催化剂的4.58、2.40倍。由此可见,CdS@g-C3N4复合核壳结构具有明显提高的可见光催化活性。
图 12是反应温度为180 ℃、反应时间为4 h,改变g-C3N4的加入量制备的CdS@g-C3N4复合核壳结构的光催化性能。结果表明,随着g-C3N4加入量的增加,其光催化性能先增大后减小。当CdS与g-C3N4的质量比约为1.9:1时,光催化性能最佳,降解率为95.2%。
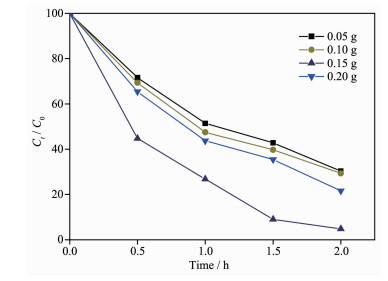
增大g-C3N4加入量,能给内核CdS提供更多的外“壳”g-C3N4,有利于形成更多的CdS@g-C3N4复合核壳结构。但继续增大g-C3N4加入量,大量外“壳”g-C3N4所产生的空间位阻效应抑制了CdS@g-C3N4复合核壳结构的生成,同时阻塞了CdS@g-C3N4核壳间隙,阻碍了光的入射和吸收,不利于光催化的进行,导致其光催化活性先增大后减小。
图 13是CdS@g-C3N4复合核壳结构光催化降解RhB的循环实验结果。由图可知,CdS@g-C3N4经3次重复实验后其光催化活性几乎不变,表明其具有良好的光催化稳定性。
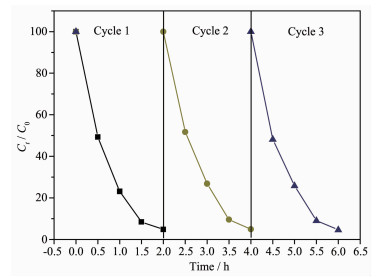
图 14考察了CdS@g-C3N4循环使用3次后的XRD、FTIR、N2吸附-脱附等温线和表观形貌。从图 14可看出,循环使用3次后,CdS@g-C3N4的晶体结构、官能团、形貌没有发生明显改变,其比表面积为55 m2·g-1,与新鲜样品相比变化很小。以上结果进一步表明,CdS@g-C3N4复合核壳结构具有良好的光催化稳定性。
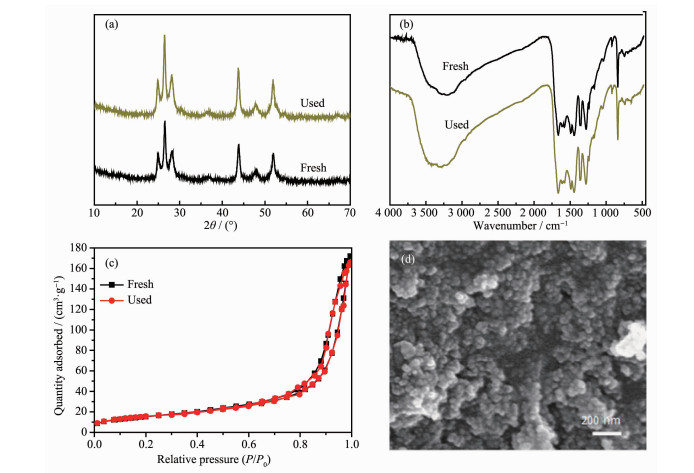
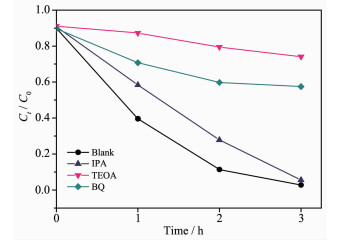
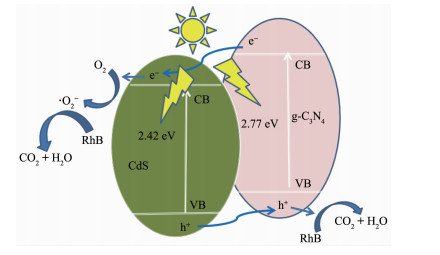
图 15考察了不同捕获剂对CdS@g-C3N4光催化性能的影响,通过引入0.01 mol·L-1的异丙醇(IPA)、三乙醇胺(TEOA)和对苯醌(BQ),相应捕获反应过程中产生的羟基自由基(HO·)、空穴(hVB+)和超氧自由基(·O2-)[44-45],以研究CdS@g-C3N4光催化降解RhB的机理。由图可知,将TEOA或BQ加入反应体系后,RhB光降解活性被明显抑制,而将IPA加入反应体系后,RhB光降解活性仅略微下降。该结果表明,空穴(hVB+)和超氧自由基(·O2-)是光催化反应体系中的主要活性物种。
CdS@g-C3N4的可见光催化机理如图 16所示。基于能级匹配原理[46],所构建的CdS/g-C3N4异质结在热力学条件有利于光生电子的产生和转移[47-48]。在可见光作用下,g-C3N4导带(CB)上的光生电子可以直接转移到CdS的CB上;同时,CdS价带(VB)上的空穴可以直接转移到g-C3N4的VB上。这促进了电子-空穴对的有效分离,提升了载流子分离效率,提高了光催化活性。此外,光生电子将吸附在催化剂表面的O2还原成·O2-,·O2-可有效地把RhB降解成CO2和H2O等小分子;同时,g-C3N4表面的电子和CdS表面的空穴也会直接或间接参与降解有机污染物RhB[49]。
采用水热法制备了CdS@g-C3N4复合材料,XRD、HRTEM、XPS、FTIR表征结果说明CdS@g-C3N4是以片状g-C3N4包覆CdS纳米颗粒形成的核壳结构,具有比纯CdS高14.0倍的比表面积。CdS与g-C3N4通过共轭效应和静电相互作用,具有明显的异质界面,可有效促进光生电子-空穴分离和电荷转移。CdS与g-C3N4复合后明显淬灭了CdS荧光,提高了CdS光电流响应值,降低了电荷转移电阻,改善了催化剂光降解RhB的活性和稳定性。空穴和超氧自由基是光催化反应体系中的主要活性物种。CdS@g-C3N4核壳结构实现了片层g-C3N4对CdS纳米颗粒的有效修饰和全方位保护,提高了CdS的光催化活性,避免了CdS团聚和失活,为构筑高效稳定的CdS基复合材料提供了参考方法。
赵晓华, 苏帅, 武广利, 等.无机化学学报, 2017, 33(2):276-284 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20170211ZHAO Xiao-Hua, SU Shuai, WU Guang-Li, et al. Chinese J. Inorg. Chem., 2017, 33(2):276-284 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20170211
Su J Z, Zhang T, Wang L, et al. Chin. J. Catal., 2017, 38(3):489-497
Wang Z Y, Chen J X, Zhang F A, et al. Rare Met. Mater. Eng., 2008, 37(2):330-333
Liu L, Hu P R, Cui W Q, et al. Int. J. Hydrogen Energy., 2017, 42(27):17435-17445 doi: 10.1016/j.ijhydene.2017.02.171
LANG Di(郎笛). Thesis for the Doctorate of Huazhong Agricultural University(华中农业大学博士论文). 2016.
Li X Y, Peng K. Appl. Clay Sci., 2018, 165:188-196 doi: 10.1016/j.clay.2018.08.017
潘金波, 刘建军, 马贺成, 等.无机化学学报, 2018, 34(8):1421-1429 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180803&flag=1PAN Jin-Bo, LIU Jian-Jun, MA He-Cheng, et al. Chinese J. Inorg. Chem., 2018, 34(8):1421-1429 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180803&flag=1
王晓雪, 高建平, 赵瑞茹, 等.无机化学学报, 2018, 34(6):1059-1064 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180607&flag=1WANG Xiao-Xue, GAO Jian-Ping, ZHAO Rui-Ru, et al. Chinese J. Inorg. Chem., 2018, 34(6):1059-1064 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180607&flag=1
李曹龙, 赵宇婷, 曹菲, 等.无机化学学报, 2013, 29(12):2535-2542 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=130493LI Cao-Long, ZHAO Yu-Ting, CAO Fei, et al. Chinese J. Inorg. Chem., 2013, 29(12):2535-2542 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=130493
李娜, 王茗, 赵北平, 等.无机化学学报, 2016, 32(6):1033-1040 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20160614&flag=1LI Na, WANG Ming, ZHAO Bei-Ping, et al. Chinese J. Inorg. Chem., 2016, 32(6):1033-1040 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20160614&flag=1
Wang D K, Zeng H, Xiong X, et al. Sci. Bull. https://doi.org/10.1016/j.scib.2019.10.015.
Zhou G, Zheng L L, Wang D K, et al. Chem. Commun., 2019, 55:4150-4153 doi: 10.1039/C9CC01161G
Wang D K, Ye P, Li K L, et al. Appl. Catal. B, 2020, 260:118182-118191 doi: 10.1016/j.apcatb.2019.118182
Wang D K, Li X, Zheng L L, et al. Nanoscale, 2018, 10:19509-19516 doi: 10.1039/C8NR06691D
Yin C C, Cui L F, Pu T T, et al. Appl. Surf. Sci., 2018, 456:464-472 doi: 10.1016/j.apsusc.2018.06.137
Li Y, Wei X, Li H, et al. RSC Adv., 2015, 5(19):14074-14080 doi: 10.1039/C4RA14690E
王智平, 赵静, 王克振.太阳能学报, 2013, 34(8):1311-1316 doi: 10.3969/j.issn.0254-0096.2013.08.002WANG Zhi-Ping, ZHAO Jing, WANG Ke-Zhen. Acta Energiae Solaris Sinica, 2013, 34(8):1311-1316 doi: 10.3969/j.issn.0254-0096.2013.08.002
申倩倩, 田赟, 薛晋波, 等.无机化学学报, 2014, 30(8):1839-1844 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20130411SHEN Qian-Qian, TIAN Bin, XUE Jin-Bo, et al. Chinese J. Inorg. Chem., 2014, 30(8):1839-1844 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20130411
Chen D Y, Ji G, Ma Y, et al. ACS Appl. Mater. Interfaces, 2011, 3(8):3078-3083 doi: 10.1021/am200592r
Wang H Y, Gong R, Qian X L. J. Membr. Sci., 2018, 8(1):14-21
Kumar S, Baruah A, Tonda S, et al. Nanoscale, 2014, 6:4830-4843 doi: 10.1039/c3nr05271k
王益林, 黄小凤, 陆建平.广西大学学报:自然科学版, 2007, 32(2):147-149, 162WANG Yi-Lin, HUANG Xiao-Feng, LU Jian-Ping. Journal of Guangxi University:Natural Science Edition, 2007, 32(2):147-149, 162
鲍淑芬.辽宁大学学报, 2010, 37(2):148-151 doi: 10.3969/j.issn.1000-5846.2010.02.014BAO Shu-Fen. Journal of Liaoning University, 2010, 37(2):148-151 doi: 10.3969/j.issn.1000-5846.2010.02.014
Fu J, Chang B B, Tian Y L, et al. J. Mater. Chem. A, 2013, 1:3083-3090 doi: 10.1039/c2ta00672c
Yang S B, Gong Y J, Zhang J S, et al. Adv. Mater., 2013, 25:2452-2456 doi: 10.1002/adma.201204453
Dong F, Wu L W, Sun Y J, et al. J. Mater. Chem., 2011, 21:15171-15174 doi: 10.1039/c1jm12844b
Muhammad A, Li Q Y, Yao J C, et al. J. Environ. Chem. Eng., 2017, 5(6):5358-5368 doi: 10.1016/j.jece.2017.10.024
Zhang S L, Hang N T, Zhang Z J, et al. J. Nanomater., 2017, 7(1):76-86
张丽, 粱青满, 戴超华, 等.中国有色金属学报, 2018, 28(7):1335-1342ZHANG Li, LIANG Qing-Man, DAI Chao-Hua, et al. The Chinese Journal of Nonferrous Metals, 2018, 28(7):1335-1342
高宁, 郭范, 郑伟威, 等.人工晶体学报, 2005, 4:632-636GAO Ning, GUO Fan, ZHENG Wei-Wei, et al. Journal of Synthetic Crystals, 2005, 4:632-636
李娇娇, 赵卫峰, 张改, 等.高等学校化学学报, 2018, 39(12):2719-2724 doi: 10.7503/cjcu20180460LI Jiao-Jiao, ZHAO Wei-Feng, ZHANG Gai, et al. Chem. J. Chin. Univ., 2018, 39(12):2719-2724 doi: 10.7503/cjcu20180460
Li Z, Zhou Z, Ma J W, et al. Appl. Catal. B, 2018, 237:288-294 doi: 10.1016/j.apcatb.2018.05.087
Zou S, Fu Z H, Xiang C, et al. Chin. J. Catal., 2015, 36(7):1077-1085 doi: 10.1016/S1872-2067(15)60827-0
张文康, 梁燕艺, 刘瑶, 等.广西科技大学学报, 2018, 29(4):66-73ZHANG Wen-Kang, LIANG Yan-Yi, LIU Yao, et al. Journal of Guangxi University of Science and Technology, 2018, 29(4):66-73
Zhang H G, Fen L J, Li C H, et al. J. Fuel Cell Sci., 2018, 46(7):871-878
梁红玉, 邹赫, 胡绍争, 等.分子催化, 2018, 32(2):152-162LIANG Hong-Yu, ZOU He, HU Shao-Zheng, et al. Journal of Molecular Catalysis, 2018, 32(2):152-162
姜建刚, 任文艺, 李霞, 等.半导体光电, 2019, 40(1):82-87JIANG Jian-Gang, REN Wen-Yi, LI Xia, et al. Semiconductor Optoelectornics, 2019, 40(1):82-87
Chong B, Chen L, Han D Z, et al. Chin. J. Catal., 2019, 40(6):959-968
Liu X M, Huang W Y, Huang G X, et al. Ceram. Int., 2015, 41:11710-11718
Yu H, Chen F Y, Chen F, et al. Appl. Surf. Sci., 2015, 6:86-91
展红全, 邓册, 吴传琦, 等.人工晶体学报, 2019, 48(2):190-195ZHAN Hong-Quan, DENG Ce, WU Chuan-Qi, et al. Journal of Synthetic Crystals, 2019, 48(2):190-195
Bai Z M, Yan X Q, Li Y, et al. Adv. Energy Mater., 2016, 6(3):1459-1501
熊剑, 何禄英.真空科学与技术学报, 2017, 37(12):1160-1165XIONG Jian, HE Lu-Ying. Chinese Journal of Vacuum Science and Technology, 2017, 37(12):1160-1165
Cui Y J, Ding Z X, Liu P. Phys. Chem. Chem. Phys., 2012, 14(4):1455-1462
Wang X, Maeda K, Thomas A. Nat. Mater., 2009, 8(1):76-80
Liu X M, Jiang Y, Guo W M, et al. Chem. Eng. J., 2013, 5(7):466-474
周文君, 沈伯雄, 张芹, 等.燃料化学学报, 2019, 47(2):249-256ZHOU Wen-Jun, SHEN Bo-Xiong, ZHANG Qin, et al. Journal of Fuel Chemistry and Technology, 2019, 47(2):249-256
郝彦忠, 曹寅虎, 孙宝, 等.化学学报, 2012, 70(10):1139-1144HAO Yan-Zhong, CAO Yin-Hu, SUN Bao, et al. Acta Chim. Sinica, 2012, 70(10):1139-1144
Long J, Liu H L, Wu S J, et al. ACS Catal., 2013, 3(4):647-654

图 1 CdS@g-C3N4复合核壳纳米微粒的制备示意图
Figure 1 Graphical illustration for the synthesis of CdS@g-C3N4 core-shell structure composite

图 2 (a) g-C3N4、CdS和CdS@g-C3N4的XRD图; (b) CdS和CdS@g-C3N4的(002)晶面衍射峰放大图
Figure 2 (a) XRD patterns of g-C3N4, CdS and CdS@g-C3N4 and (b) Enlarged detail of (002) peak of CdS and CdS@g-C3N4

图 3 (a) g-C3N4、(b) CdS和(c) CdS@g-C3N4的SEM图
Figure 3 SEM images of (a) g-C3N4, (b) CdS and (c) CdS@g-C3N4

图 7 g-C3N4、CdS和CdS@g-C3N4的FT-IR谱图
Figure 7 FT-IR spectra of g-C3N4, CdS and CdS@g-C3N4

图 8 g-C3N4、CdS和CdS@g-C3N4的N2吸附-脱附等温线
Figure 8 N2 adsorption-desorption isotherms of g-C3N4, CdS and CdS@g-C3N4

图 9 g-C3N4、CdS和CdS@g-C3N4的(a) UV-Vis DRS和(b) PL光谱图
Figure 9 (a) UV-Vis DRS and (b) PL spectra of g-C3N4, CdS and CdS@g-C3N4

图 10 g-C3N4、CdS和CdS@g-C3N4在可见光照射下的(a) I-t和(b) EIS曲线
Figure 10 I-t (a) and EIS (b) curves of g-C3N4, CdS and CdS@g-C3N4

图 11 g-C3N4, CdS和CdS@g-C3N4在可见光照下(a)降解RhB的曲线和(b)一级动力学曲线
Figure 11 (a) Photocatalytic degradation and (b) First-order kinetic curves of RhB by g-C3N4, CdS and CdS@g-C3N4 under visible light

图 12 g-C3N4添加量对CdS@g-C3N4可见光照下降解RhB的性能影响
Figure 12 Effect of the feed amount of g-C3N4 on the photocatalytic degradation of RhB over CdS@g-C3N4 under visible light irradiation

图 13 CdS@g-C3N4复合材料在可见光照下降解RhB的循环实验结果
Figure 13 Cyclic test results for the photocatalytic degradation of RhB by CdS@g-C3N4 under visible light irradiation

图 14 光催化循环3次前后CdS@g-C3N4的(a) XRD图、(b) FTIR图谱、(c)N2吸附-脱附等温线; (d)循环使用后的SEM图
Figure 14 (a) XRD patterns, (b) FT-IR spectra and (c) N2 adsorption-desorption isotherms of fresh CdS@g-C3N4 and the used after the 3rd run for photocatalysis; (d) SEM image of the used CdS@g-C3N4

图 15 不同捕获剂对CdS@g-C3N4光催化性能的影响
Figure 15 Effect of different scavengers on the RhB degradation in the presence of CdS@g-C3N4 heterojunction under visible light

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:



