-
[1]
Lim B, Jiang M J, Camargo P H C, et al. Science, 2009, 324(5932):1302-1305 doi: 10.1126/science.1170377
-
[2]
Yu T, Kim D Y, Zhang H, et al. Angew. Chem. Int. Ed., 2011, 50(12):2773-2777 doi: 10.1002/anie.201007859
-
[3]
Lu Y C, Xu Z C, Gasteiger H A, et al. J. Am. Chem. Soc., 2010, 132(35):12170-12171 doi: 10.1021/ja1036572
-
[4]
Lee Y, Suntivich J, May K J, et al. J. Phys. Chem. Lett., 2012, 3(3):399-404 doi: 10.1021/jz2016507
-
[5]
Gasteiger H A, Kocha S S, Sompalli B, et al. Appl. Catal. B, 2005, 56(1/2):9-35 doi: 10.1016/j.apcatb.2004.06.021
-
[6]
Li Y G, Dai H J. Chem. Soc. Rev., 2014, 43(15):5257-5275 doi: 10.1039/C4CS00015C
-
[7]
Liang Y Y, Li Y G, Wang H L, et al. Nat. Mater., 2011, 10(10):780-786 doi: 10.1038/nmat3087
-
[8]
Wang Z L, Xu D, Xu J J, et al. Chem. Soc. Rev., 2014, 43(22):7746-7786 doi: 10.1039/C3CS60248F
-
[9]
Zhao Y F, Chen S Q, Sun B, et al. Sci. Rep., 2015, 5:7629 doi: 10.1038/srep07629
-
[10]
Chen C F, King G, Dickerson R M, et al. Nano Energy, 2015, 13:423-432 doi: 10.1016/j.nanoen.2015.03.005
-
[11]
Prabu M, Ramakrishnan P, Shanmugam S. Electrochem. Commun., 2014, 41:59-63 doi: 10.1016/j.elecom.2014.01.027
-
[12]
Liu Y L, Higgins D C, Wu J, et al. Electrochem. Commun., 2013, 34:125-129 doi: 10.1016/j.elecom.2013.05.035
-
[13]
Wang L, Zhao X, Lu Y H, et al. J. Electrochem. Soc., 2011, 158(12):A1379-A1382 doi: 10.1149/2.068112jes
-
[14]
Fu J, Hassan F M, Li J D, et al. Adv. Mater., 2016, 28(30):6421-6428 doi: 10.1002/adma.201600762
-
[15]
Li G, Wang X L, Fu J, et al. Angew. Chem. Int. Ed., 2016, 55(16):4977-4982 doi: 10.1002/anie.201600750
-
[16]
Liu X, Liu W, Ko M, et al. Adv. Funct. Mater., 2015, 25(36):5799-5808 doi: 10.1002/adfm.201502217
-
[17]
Biddinger E J, von Deak D, Ozkan U S. Top. Catal., 2009, 52(11):1566-1574 doi: 10.1007/s11244-009-9289-y
-
[18]
Matter P H, Wang E, Arias M, et al. J. Mol. Catal. A:Chem., 2007, 264(1/2):73-81 doi: 10.1016/j.molcata.2006.09.008
-
[19]
Subramanian N P, Li X G, Nallathambi V, et al. J. Power Sources, 2009, 188(1):38-44 doi: 10.1016/j.jpowsour.2008.11.087
-
[20]
Li Q, Cao R, Cho J, et al. Adv. Energy Mater., 2014, 4(6):1301415 doi: 10.1002/aenm.201301415
-
[21]
Paraknowitsch J P, Thomas A. Energy Environ. Sci., 2013, 6(10):2839-2855 doi: 10.1039/c3ee41444b
-
[22]
Qu L T, Liu Y, Baek J B, et al. ACS Nano, 2010, 4(3):1321-1326 doi: 10.1021/nn901850u
-
[23]
Higgins D, Chen Z, Lee D U, et al. J. Mater. Chem. A, 2013, 1(7):2639-2645 doi: 10.1039/c2ta00944g
-
[24]
Geng D S, Chen Y, Chen Y G, et al. Energy Environ. Sci., 2011, 4(3):760-764
-
[25]
Lin Z Y, Song M K, Ding Y, et al. Phys. Chem. Chem. Phys., 2012, 14(10):3381-3387 doi: 10.1039/c2cp00032f
-
[26]
Dhavale V M, Gaikwad S S, Kurungot S. J. Mater. Chem. A, 2014, 2(5):1383-1390 doi: 10.1039/C3TA14005A
-
[27]
Yuan P F, Sun Q, Jia Y, et al. J. Phy. Chem. C, 2014, 118(32):18315-18324 doi: 10.1021/jp502493g
-
[28]
Liu C S, Liu X C, Wang G C. et al. J. Electroanal. Chem., 2014, 728:41-50 doi: 10.1016/j.jelechem.2014.06.024
-
[29]
Duan J J, Chen S, Dai S, et al. Adv. Funct. Mater., 2014, 24(14):2072-2078 doi: 10.1002/adfm.201302940
-
[30]
Liang Y Y, Li Y G, Wang H L, et al. Nat. Mater., 2011, 10(10):780-786 doi: 10.1038/nmat3087
-
[31]
Li S S, Cong H P, Wang P, et al. Nanoscale, 2014, 6(13):7534-7541 doi: 10.1039/C4NR02101K
-
[32]
Bag S, Roy K, Gopinath C S, et al. ACS Appl. Mater. Interfaces, 2014, 6(4):2692-2699 doi: 10.1021/am405213z
-
[33]
Ayala P, Arenal R, Rummeli M, et al. Carbon, 2010, 48(3):575-586 doi: 10.1016/j.carbon.2009.10.009
-
[34]
Glerup M, Castignolles M, Holzinger M, et al. Chem. Commun., 2003(20):2542-2543 doi: 10.1039/b303793b
-
[35]
Xiong C, Wei Z D, Hu B S, et al. J. Power Sources, 2012, 215:216-220 doi: 10.1016/j.jpowsour.2012.04.057
-
[36]
Lin J H, Chen C S, Zeng Z Y, et al. Nanoscale, 2012, 4(15):4757-4764 doi: 10.1039/c2nr30854a
-
[37]
Zeng M, Liu Y L, Zhao F P, et al. Adv. Funct. Mater., 2016, 26(24):4397-4404 doi: 10.1002/adfm.201600636
-
[38]
Higgins D, Chen Z, Lee D U, et al. J. Mater. Chem. A, 2013, 1(7):2639-2645 doi: 10.1039/c2ta00944g
-
[39]
Kim H, Lee K, Woo S I, et al. Phys. Chem. Chem. Phys., 2011, 13(39):17505-17510 doi: 10.1039/c1cp21665a
-
[40]
Lai L F, Potts J R, Zhan D, et al. Energy Environ. Sci., 2012, 5(7):7936-7942 doi: 10.1039/c2ee21802j
-
[41]
Sharifi T, Hu G, Jia X E, et al. ACS Nano, 2012, 6(10):8904-8912 doi: 10.1021/nn302906r
-
[42]
Zhang X, Liu R R, Zang Y P, et al. Chem. Commun., 2016, 52(35):5946-5949 doi: 10.1039/C6CC02513G
-
[43]
Lee D U, Park M G, Park H W, et al. ChemSusChem., 2015, 8(18):3129-3138 doi: 10.1002/cssc.201500609
-
[44]
Wang Z J, Li B, Ge X M, et al. Small, 2016, 12(19):2580-2587 doi: 10.1002/smll.201503694
-
[45]
Guo L G, Ding Y, Qin C Q, et al. Electrochim. Acta, 2016, 187:234-242 doi: 10.1016/j.electacta.2015.11.065
-
[46]
Gong K P, Du F, Xia Z H, et al. Science, 2009, 323(5915):760-764 doi: 10.1126/science.1168049
-
[47]
Liu Y T, Li K X, Liu Y, et al. J. Mater. Chem. A, 2015, 3(42):21149-21158 doi: 10.1039/C5TA04595A
-
[48]
Wu J, Yang Z R, Sun Q J, et al. Electrochim. Acta, 2014, 127:53-60 doi: 10.1016/j.electacta.2014.02.016
-
[49]
Zhao N N, Nie W, Mao J, et al. Small, 2010, 6(22):2558-2565 doi: 10.1002/smll.201001218
-
[50]
Li Y G, Wang H L, Xie L M, et al. J. Am. Chem. Soc., 2011, 133(49):7296-7299 doi: 10.1021/ja201269b
-
[51]
Liang Y Y, Wang H L, Casalongue H S, et al. Nano Res., 2010, 3(10):701-705 doi: 10.1007/s12274-010-0033-5
-
[52]
Yang Y, Li J X, Wang H, et al. Angew. Chem. Int. Ed., 2011, 50(9):2148-2150 doi: 10.1002/anie.201005941
-
[53]
Jiang Z Q, Jiang Z J, Maiyalagan T, et al. J. Mater. Chem. A, 2016, 4(16):5877-5899 doi: 10.1039/C6TA01349J
-
[54]
Liu Q, Wang Y B, Dai L M, et al. Adv. Mater., 2016, 28(15):3000-3006 doi: 10.1002/adma.201506112
-
[55]
Zeng M, Liu Y L, Zhao F P, et al. Adv. Funct. Mater., 2016, 26(24):4397-4404 doi: 10.1002/adfm.201600636


 下载:
下载:

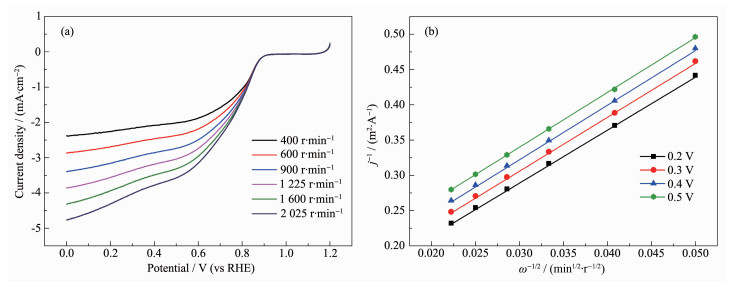
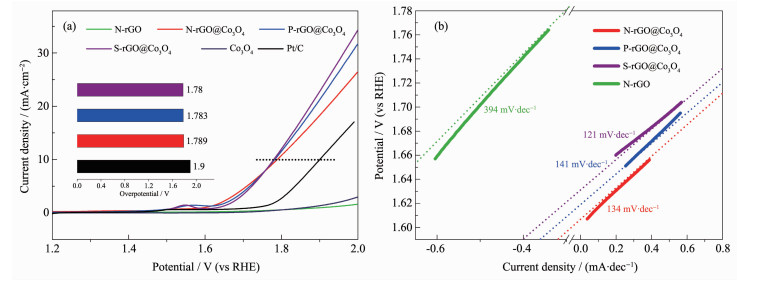
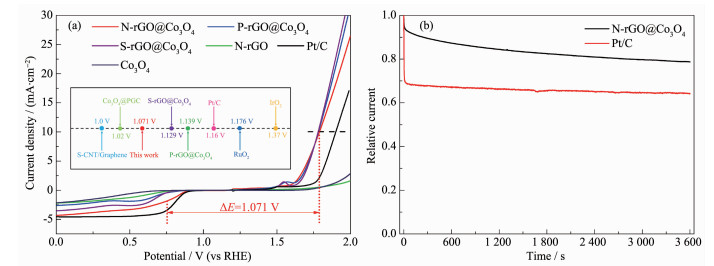

 下载:
下载:










