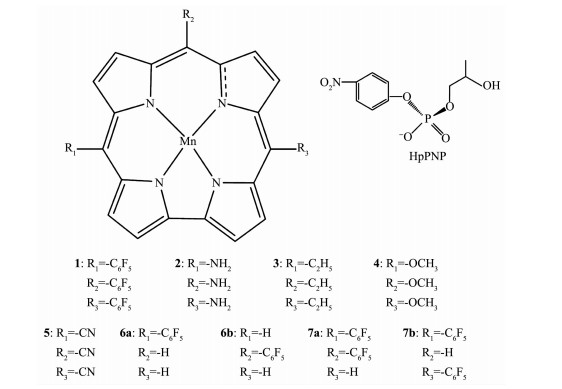
图 1.
咔咯锰(Ⅲ)配合物结构
Figure 1.
Structures of corrole manganese(Ⅲ) complexes

咔咯是一类具有18π电子共轭体系的类卟啉大环化合物[1],环中有三个亚胺基团,比较容易进行酸式解离反应,形成三价阴离子配体,其空腔小能与金属离子形成稳定的高价咔咯金属配合物[2-6]。实验研究发现金属咔咯配合物能够与DNA相互作用并催化断裂DNA,在光动力治疗和抗肿瘤等生物医学应用方面具有良好前景,咔咯金属配合物和DNA的相互作用及其核酸酶活性逐渐成为生物化学领域的热点研究之一[7-15]。咔咯锰(Ⅲ)配合物的活性中心锰离子具有良好的氧化还原特性,且结构稳定,因此在结合DNA、催化氧化DNA断裂等方面具有较好的化学生物活性[7]。但是其断裂位置以及机理仍无法从现有实验研究中得到很好的解答。
磷酸酯及其衍生物广泛地存在于各种生物分子中,其动力学稳定性在遗传信息的储存和代谢过程中发挥关键的作用[16]。磷酸二酯结构为DNA和RNA骨架的重要组成结构,在DNA和RNA核苷酸之间的磷酸二酯键可以被催化氧化裂解或水解[17]。DNA、RNA的复制和翻译都与磷酸酯的裂解密切相关,因此深入了解磷酸二酯的水解机理具有重要的意义。一般认为在水解过程中,亲核分子与体系的磷中心原子之间的亲核进攻过程会使离去基团P-O键断裂并产生解离[18-19]。其反应过程主要有一般碱催化(GBC)和特殊碱催化(SBC)两种机理[20-23]。在实际研究中,选用RNA磷酸二酯的类似物2-羟丙基-4-硝基苯基磷酸酯(HpPNP)作为磷酸二酯结构的参照物的研究较多[17, 20-21, 24]。因此,我们从理论上研究咔咯锰(Ⅲ)金属配合物催化RNA磷酸二酯类似物HpPNP的水解断裂反应,探究其水解断裂机理及中位取代基性质对反应的影响,从分子水平上了解这类化合物水解断裂磷酸二酯键链的作用机制。
首先以5,10,15-三(五氟苯基)咔咯锰(TPFC)配合物1为模型,研究其催化HpPNP水解反应机理,然后在咔咯环上的中位5、10和15同时引入吸电子基团(-CN、-OCH3)和供电子基团(-NH2、-CH2CH3)构成4个配合物系列(2~5)研究取代基性质对反应的影响;另一方面再以中位被-C6F5取代,构成一取代、二取代和三取代等5个配合物(1,6a~7b)研究取代基数量对反应的影响,配合物的结构见图 1。我们课题组参考以往文献发现B3LYP[25]方法优化的结构更接近实验值[26],因此本文采用密度泛函理论的B3LYP方法,锰离子用LanL2DZ基组,磷原子用6-311G*基组,C、N、H、O、F用6-31G*基组。有关理论计算显示咔咯锰(Ⅲ)配合物五重态最稳定[26],因此本文所有配合物分子均为五重态。各反应物、生成物优化构型的振动频率均无虚频,表明分子构型稳定,各过渡态化合物有且仅有一个虚频, 并经IRC验证,获得相应的反应物和产物结构。以上所有的计算过程均使用Gaussian09[27]程序完成。
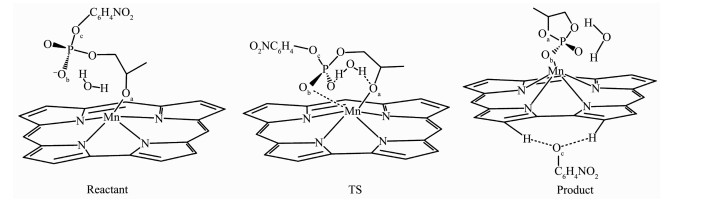
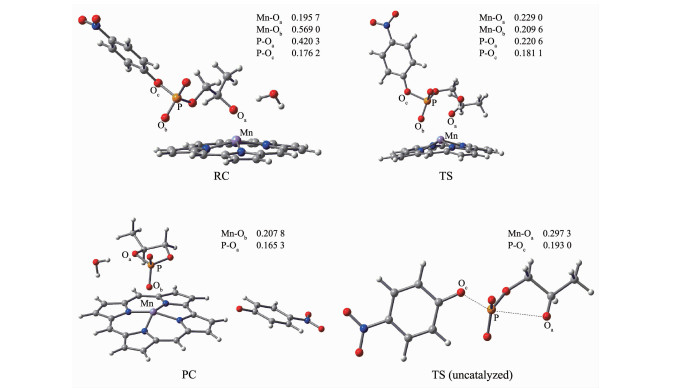
经过对咔咯锰(Ⅲ)配合物催化HpPNP水解反应过渡态构型的多次尝试计算,找出在OH-作用下以SBC机理进行水解反应的过渡态TS,并通过IRC方法得到反应物和产物结构,获得磷酸二酯类似物HpPNP的一条可能水解途径,如图 2所示。以5,10,15-三(五氟苯基)咔咯锰(TPFC)配合物1为例,在过渡态(TS)时,水分子与Oa和磷酰基团上的O原子形成双氢键结构。在水解过程中(见图 3),锰离子与进攻的Oa原子之间的配位键长从0.195 7 nm增加到0.229 0 nm,锰离子与Ob原子之间的距离从0.569 0 nm减少到0.209 6 nm,Mn与Oa、Ob原子形成双配位结构,该结构能稳定过渡态的构型。P原子与进攻的Oa原子之间的距离从0.420 3 nm减少到0.220 6 nm,P原子与离去的Oc原子之间的键长由0.176 2 nm增加到0.181 1 nm。随着水解反应继续进行, 最终P-Oc键断裂,P-Oa键形成,键长为0.165 3 nm,Mn-Ob形成有效配位键长0.207 8 nm。表明在咔咯锰(III)配合物催化作用下P-Oa产生键合作用,削弱P-Oc键。无催化剂中性条件下的过渡态如图 3中TS(uncatalyzed),与配合物1中的TS对比,P原子与进攻的Oa原子之间的距离从0.297 3 nm减少到0.220 6 nm,P原子与离去的Oc原子之间的键长由0.193 0 nm减少到0.181 1 nm,说明在催化剂的作用下,过渡态从一个松散的结构到一个紧凑的结构。
电荷转移在化学反应中也是基本的过程之一,根据NPA(自然电荷布局分析),各反应态的平衡构型的重要原子或基团的电荷列于表 1,随着反应的进行,HpPNP中作为离去的基团-O-C6H4-NO2带的负电荷增加,从-0.665至-0.692,离去时增至-0.968,咔咯锰的负电荷从-0.527减至-0.198,说明水解反应过程中负电荷从咔咯锰转移到离去基团-O-C6H4-NO2上面,导致离去基团容易解离,P-Oc键断裂。
 下载:
导出CSV
下载:
导出CSV
| RC | TS | PC | |
| Q-O-C6H4-NO2 | -0.665 | -0.692 | -0.968 |
| Q Corrole Mn | -0.527 | -0.308 | -0.198 |
通过计算HpPNP分别在无催化剂中性条件和咔咯锰(Ⅲ)配合物1催化条件下的水解反应能垒,以反应物优化得到的零点校正能为零势能面,得出HpPNP水解反应能垒(Supporting information)。咔咯锰(Ⅲ)配合物1催化HpPNP水解的反应能垒为77.23 kJ·mol-1,明显低于无催化剂时的计算水解反应能垒96.05 kJ·mol-1和实验值118.30 kJ·mol-1 [17]。同时,也低于文献报道Zn(OH):[12]aneN3配合物催化HpPNP水解的计算反应能垒92.39 kJ·mol-1 [24]。这说明咔咯锰(Ⅲ)配合物能明显降低HpPNP水解的反应能垒,加快反应速率。
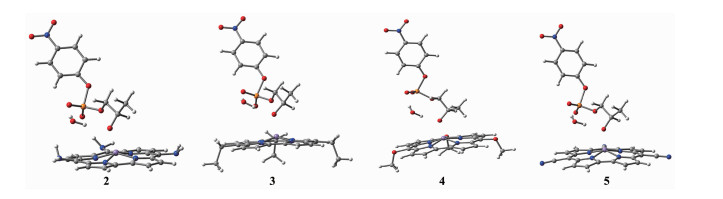
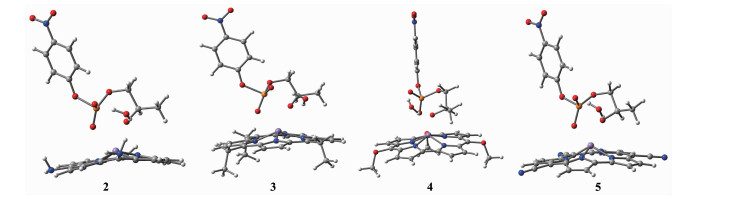
在咔咯环上的中位引入不同的吸电子基团(-CN、-OCH3)和供电子基团(-NH2、-CH2CH3)构成4个配合物,考察中位取代基对水解反应的影响。对咔咯锰(Ⅲ)中位不同取代基的反应物1~5的结构进行几何优化,优化结构如图 4所示,从图中可以看出反应物结构相似。优化的部分几何结构参数列于表 2,从表中可见,随着取代基吸电子能力的增强,Mn-Oa键长呈规律性缩短,从0.200 6 nm减少到0.193 6 nm,其它结构参数并没有发生明显变化,说明反应前中位取代基对反应物构型影响并不明显。
White: hydrogen, Gray: carbon, Blue: nitrogen, Purple: manganese, Red: oxygen, Orange: phosphorus
下载:
导出CSV
| Complex | Substituent | d(Mn-Oa)/nm | d(Mn-Ob)/nm | d(P-QC)/nm | d(P-Qa)/nm |
| 2 | -NH2 | 0.200 6 | 0.579 9 | 0.176 3 | 0.422 3 |
| 3 | -C2H5 | 0.197 6 | 0.586 6 | 0.177 6 | 0.420 8 |
| 4 | -OCH3 | 0.195 8 | 0.648 3 | 0.178 1 | 0.424 6 |
| 1 | -C6F5 | 0.195 7 | 0.569 0 | 0.176 2 | 0.420 3 |
| 5 | -CN | 0.193 6 | 0.576 4 | 0.176 5 | 0.419 7 |
计算中位不同取代基的咔咯锰(Ⅲ)配合物催化HpPNP水解反应的过渡态,结构见图 5。各配合物的过渡态结构相似,形成双氢键结构和双配位结构。几何结构优化所得参数见表 3,随着取代基的吸电子性能增加,Mn-Ob的键长呈规律性缩短,从0.236 0 nm减少到0.196 2 nm,进攻基团的P-Oa键长从0.235 1 nm逐渐减少至0.216 1 nm,过渡态结构越来越紧凑。
White: hydrogen, Gray: carbon, Blue: nitrogen, Purple: manganese, Red: oxygen, Orange: phosphorus
下载:
导出CSV
| Complex | Substituent | d(Mn-Oa) / nm | d(Mn-Ob) / nm | d(P-Oc) / nm | d(P-Oa) / nm |
| 2 | -NH2 | 0.231 2 | 0.236 0 | 0.183 0 | 0.235 1 |
| 3 | -C2H5 | 0.225 5 | 0.231 4 | 0.184 0 | 0.228 3 |
| 4 | -OCH3 | 0.225 8 | 0.224 7 | 0.183 7 | 0.225 0 |
| 5 | -CN | 0.218 3 | 0.196 2 | 0.178 0 | 0.216 1 |
对各配合物的反应态和过渡态进行二级微扰稳定化能分析(表 4),与反应态相比,过渡态P与Oc间的E(2)有不同程度的下降,进攻基团P与Oa间的 E(2)值大于190 kJ·mol-1且E(2)值不断增加,随着取代基吸电子能力的增强,P与Oa间的作用逐渐增大。为进一步探讨取代基对配合物前线轨道能级的影响,对各个配合物的前线分子轨道进行分析,具体数据见表 5。从表中可以看出,取代基的吸电子效应使体系各轨道能下降,其中占据轨道能下降明显,HOMO轨道主要分布在咔咯骨架上,LUMO轨道主要分布在离去基团上(轨道图见Supporting information)。
下载:
导出CSV
| Complex | 2 | 3 | 4 | 1 | 5 |
| RC P-Oc | 1 143.06 | 1 104.57 | 1 082.79 | 1 242.30 | 1 195.27 |
| TS P-Oc | 1 045.30 | 1 072.09 | 1 066.86 | 1 115.02 | 1 145.11 |
| TS P-Oa | 194.58 | 228.44 | 250.09 | 286.96 | 304.14 |
下载:
导出CSV
| Complex | 2 | 3 | 4 | 1 | 5 |
| Substituent | -NH2 | -C2H5 | -OCH3 | -C6F5 | -CN |
| EHOMO | 0.052 2 | 0.028 7 | 0.018 8 | -0.003 9 | -0.020 3 |
| ELUMO | 0.070 9 | 0.077 8 | 0.081 9 | 0.069 2 | 0.059 0 |
对各个配合物催化HpPNP水解反应能垒进行分析,具体数据见表 6。表中数据显示随着取代基吸电子能力的增强,HpPNP水解能垒由104.81 kJ·mol-1(含-NH2配合物2)降低至63.76 kJ·mol-1(含-CN配合物5),可见吸电子取代基将显著降低反应能垒,有利于水解反应的进行。
下载:
导出CSV
| Complex | 2 | 3 | 4 | l | 5 |
| Energy barrier | 104.81 | 92.09 | 81.49 | 77.23 | 63.76 |
从反应态到过渡态,随着取代基吸电子能力的增强,P-Oc的键长增加的幅度逐渐减小,例如配合物2和5的P-Oc键长增加值分别为0.006 7和0.001 5 nm,导致体系过渡态的能量上升幅度也相应减少;同时,P-Oa的键长缩短的幅度增大,例如配合物2和5的P-Oa键长减少值分别为0.187 2和0.203 6 nm,导致体系过渡态的能量下降幅度增大。两者共同作用下,导致上述体系的反应能垒随取代基吸电子能力的增强而下降。因此在上述5个体系中,含强吸电子基-CN的配合物5催化HpPNP水解的反应能垒最低。
当中位取代基为五氟苯基时,探究取代基数目对咔咯锰(Ⅲ)配合物催化HpPNP水解的影响。对反应物1,6a~7b在五重态下的结构进行几何优化,优化结构如图 6所示,各体系优化的部分作用键长列于表 7。各键长并没有发生明显变化,说明反应前中位五氟苯基的取代数对咔咯锰(Ⅲ)配合物与HpPNP作用影响并不明显。
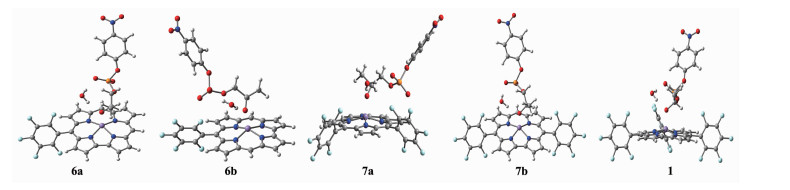
White: hydrogen, Gray: carbon, Blue: nitrogen, Purple: manganese, Red: oxygen, Orange: phosphorus
下载:
导出CSV
| Complex | d(Mn-Oa) / nm | d(Mn-Ob) / nm | d(P-Oc) / nm | d(P-Oa) / nm |
| 6a | 0.197 0 | 0.566 0 | 0.177 6 | 0.415 7 |
| 6b | 0.196 9 | 0.579 4 | 0.177 3 | 0.417 3 |
| 7a | 0.196 4 | 0.570 1 | 0.176 8 | 0.417 8 |
| 7b | 0.196 0 | 0.564 9 | 0.177 0 | 0.415 3 |
| l | 0.195 7 | 0.569 0 | 0.176 2 | 0.420 3 |
咔咯锰(Ⅲ)配合物1,6a~7b催化HpPNP水解反应的过渡态见图 7,各配合物的过渡态结构均呈双配位和双氢键结构。几何结构优化所得部分键长见表 8。从表 8可见,随着五氟苯基数目的增加,Mn-Ob键长从单(五氟苯基)咔咯锰配合物的0.223 3 nm减至双(五氟苯基)咔咯锰配合物的0.215 6 nm,而三(五氟苯基)咔咯锰配合物降至0.2096 nm。P-Oc键长从单(五氟苯基)咔咯锰配合物的0.183 5 nm减至双(五氟苯基)咔咯锰配合物的0.182 1 nm,而三(五氟苯基)咔咯锰配合物降至0.181 1 nm。过渡态结构越来越紧凑。而配合物6b和7a由于受到五氟苯基与HpPNP中苯环的排斥作用,导致P-Oa键的键长缩短。
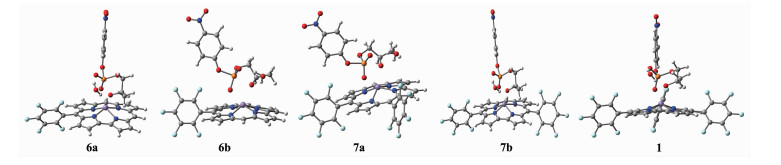
White: hydrogen, Gray: carbon, Blue: nitrogen, Purple: manganese, Red: oxygen, Orange: phosphorus
下载:
导出CSV
| Complex | d(Mn-Oa) / nm | d(Mn-Ob) / nm | d(P-Oc) / nm | d(P-Oa) / nm |
| 6a | 0.226 7 | 0.223 3 | 0.183 5 | 0.224 9 |
| 6b | 0.228 9 | 0.220 7 | 0.183 1 | 0.222 6 |
| 7a | 0.230 1 | 0.2156 | 0.182 3 | 0.221 2 |
| 7b | 0.226 4 | 0.217 3 | 0.182 1 | 0.224 7 |
| 1 | 0.229 0 | 0.209 6 | 0.181 1 | 0.220 6 |
对各配合物的反应态和过渡态进行了二级微扰稳定化能分析(表 9)。与反应态相比,过渡态P与Oc间的E(2)有不同程度的下降,进攻基团P与Oa间的E(2)值大于245 kJ·mol-1,E(2)值总体逐渐增大,而配合物7b可能是空间位阻小导致进攻基团P与Oa间的E(2)值较小,随着五氟苯基数目的增多,P与Oa间的作用总体增大。从表 10中可以看出随着吸电子取代基数目的增加,HOMO占据轨道能下降明显,LUMO轨道能没有明显变化。
下载:
导出CSV
| Complex | 6a | 6b | 7a | 7b | 1 |
| RC P-Oc | 1 102.52 | 1 106.36 | 1 126.00 | 1 122.75 | 1 242.30 |
| TS P-Oc | 1 076.22 | 1 084.30 | 1 101.43 | 1 042.12 | 1 115.02 |
| TS P-Oa | 250.38 | 264.43 | 281.23 | 247.54 | 286.96 |
下载:
导出CSV
| Complex | 6a | 6b | 7a | 7b | 1 |
| EH0M0 | 0.015 2 | 0.013 9 | 0.003 7 | 0.004 4 | -0.003 9 |
| ELUM0 | 0.076 6 | 0.078 1 | 0.074 1 | 0.074 2 | 0.069 2 |
对各个配合物催化HpPNP水解反应能垒进行分析,具体数据见表 11,发现随着五氟苯基数目的增多,反应能垒由85.74 kJ·mol-1减至77.23 kJ· mol-1,说明在咔咯锰(Ⅲ)配合物的中位引入不同数目的五氟苯基能显著降低水解反应能垒,促进水解断裂反应的进行。
下载:
导出CSV
| Complex | 6a | 6b | 7a | 7b | 1 |
| Energy barrier | 85.74 | 85.56 | 82.26 | 82.04 | 77.23 |
另一方面,上述取代基数目的变化导致反应能垒变化幅度约为8 kJ·mol-1,而吸电子取代基性质变化导致反应能垒变化幅度大大提升到41 kJ·mol-1,远大于取代基数目的变化对反应能垒的影响。理论上,在HpPNP水解反应中,咔咯锰(Ⅲ)配合物中位引入强吸电子基团能获得更低的反应能垒。
探究了咔咯锰(Ⅲ)催化HpPNP水解断裂的反应机理并探讨了中位取代基对水解反应的影响,水解的关键在于P-Oa键的形成与P-Oc键的断裂,在过渡态时水分子与Oa和磷酰基团上的O原子形成双氢键结构,Mn与Oa、Ob原子形成双配位结构。其中含-CN取代基的配合物6反应能垒最低为63.76 kJ·mol-1,为无催化剂反应能垒的66%左右,并且随着取代基吸电子能力的增强,水解反应能垒有明显降低。
Supporting information is available at http://www.wjhxxb.cn
Johnson A W, Kay I T.. J. Chem. Soc., 1965, 223:1620-1629
Kadish K M, Erben C, Ou Z, et al. Inorg. Chem., 2000, 39: 3312-3219 doi: 10.1021/ic991361m
Gross Z, Galili N, Simkhovich L, et al. Org. Lett., 1999, 1: 599-602 doi: 10.1021/ol990739h
Paolesse R, Jaquinod L, Nurco D J, et al. Chem. Commun., 1999, 30:1307-1308
Gryko D T, Jadach K.. J. Org. Chem., 2001, 66:4267-4275 doi: 10.1021/jo010146w
Ghosh A, Wondimagegn T, Parusel A B J.. J. Am. Chem. Soc., 2000, 122:5100-5104 doi: 10.1021/ja9943243
刘海洋, 刘兰英, 张雷, 等.高等学校化学学报, 2007, 28:1628-1630 doi: 10.3321/j.issn:0251-0790.2007.09.031LIU Hai-Yang, LIU Lan-Ying, ZHANG Lei, et al. Chem. J. Chinese Universities, 2007, 28:1628-1630 doi: 10.3321/j.issn:0251-0790.2007.09.031
Shi L, Liu H Y, Si L P, et al. Chin. Chem. Lett., 2010, 21: 373-375 doi: 10.1016/j.cclet.2009.11.027
Lu J, Liu H Y, Shi L, et al. Chin. Chem. Lett., 2011, 22:101- 104 doi: 10.1016/j.cclet.2010.09.005
史蕾, 江焕峰, 尹伟, 等.物理化学学报, 2012, 28:465- 469 doi: 10.3866/PKU.WHXB201111291SHI Lei, JIANG Huan-Feng, YIN Wei, et al. Acta Phys.-Chim. Sin., 2012, 28:465- 469 doi: 10.3866/PKU.WHXB201111291
Huang J T, Wang X L, Zhang Y, et al. Transition Met. Chem., 2013, 38:283-289 doi: 10.1007/s11243-013-9689-5
黄俊腾, 张阳, 王湘利, 等.无机化学学报, 2013, 29:1649-1656 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20130811&flag=1HUANG Jun-Teng, ZHANG Yang, WANG Xiang-Li, et al. Chinese J. Inorg. Chem., 2013, 29:1649-1656 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20130811&flag=1
张阳, 陈欢, 闻金燕, 等.高等学校化学学报, 2013, 34:2462-2469 doi: 10.7503/cjcu20130610ZHANG Yang, CHEN Huan, WEN Jin-Yan, et al. Chem. J. Chinese Universities, 2013, 34:2462-2469 doi: 10.7503/cjcu20130610
Zhang Y, Wang Q, Wen J Y, et al. Chin. J. Chem., 2013, 31: 1321-1328 doi: 10.1002/cjoc.201300488
Zhang Y, Wen J Y, Wang X L, et al. Appl. Organomet. Chem., 2014, 28:559-566 doi: 10.1002/aoc.3163
Zhang X, Liu X, Phillips D L, et al. ACS Catal., 2016, 6:248 -257 doi: 10.1021/acscatal.5b01735
Gao H, Ke Z, Deyonker N J, et al. J. Am. Chem. Soc., 2011, 133:2904-2915 doi: 10.1021/ja106456u
刘长林, 余四旺, 徐辉碧, 等.无机化学学报, 2000, 3:374-384 doi: 10.3321/j.issn:1001-4861.2000.03.002LIU Chang-Lin, YU Si-Wang, XUN Hui-Bi, et al. Chinese J. Inorg. Chem., 2000, 3:374-384 doi: 10.3321/j.issn:1001-4861.2000.03.002
Zhang X, Gao H, Xu H Y, et al. J. Mol. Catal. A, 2013, 368: 53-60
Feng G, Mareque J C, Torres R, et al. J. Am. Chem. Soc., 2005, 127:13470-13471 doi: 10.1021/ja054003t
Fan Y, Gao Y Q.. J. Am. Chem. Soc., 2007, 129:905-913 doi: 10.1021/ja0660251
Morrow J R, Amyes T L, Richard J P.. Acc. Chem. Res., 2008, 41:539-548 doi: 10.1021/ar7002013
Livieri M, Mancin F, Saielli G, et al. Chem.-Eur. J., 2007, 13:2246-2256 doi: 10.1002/chem.200600672
Zhang X P, Zhu Y J, Gao H, et al. Inorg. Chem., 2014, 53: 11903-11912 doi: 10.1021/ic501084a
Becke A D.. J. Chem. Phys., 1993, 98:5648-5652 doi: 10.1063/1.464913
何婧, 徐志广, 曾允秀, 等.物理化学学报, 2012, 28:1658-1664 doi: 10.3866/PKU.WHXB201205101HE Jing, XU Zhi-Guang, ZENG Yun-Xiu, et al. Acta Phys.-Chim. Sin., 2012, 28:1658-1664 doi: 10.3866/PKU.WHXB201205101
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision A.02; Gaussian Inc., Wallingford, CT, 2009.

图 2 咔咯锰(Ⅲ)配合物催化HpPNP水解反应机理简图
Figure 2 Schematic diagram of HpPNP hydrolysis reaction mechanism of corrole manganese(Ⅲ) complex

图 3 配合物1催化HpPNP水解过程中的反应物、过渡态和产物的几何优化构型及主要键长(nm)
Figure 3 Optimal configurations of reactant, transition state and product with some bond length (nm) during HpPNP hydrolysis catalyzed by complex 1

图 4 反应物2~5的几何优化结构图
Figure 4 Optimized geometrical structures of reactants 2~5
White: hydrogen, Gray: carbon, Blue: nitrogen, Purple: manganese, Red: oxygen, Orange: phosphorus

图 5 反应物2~5过渡态的几何优化结构图
Figure 5 Optimized geometrical structures of transition states of reactants 2~5
White: hydrogen, Gray: carbon, Blue: nitrogen, Purple: manganese, Red: oxygen, Orange: phosphorus

图 6 反应物1, 6a~7b的几何优化结构图
Figure 6 Optimized geometrical structures of reactants 1, 6a~7b
White: hydrogen, Gray: carbon, Blue: nitrogen, Purple: manganese, Red: oxygen, Orange: phosphorus

图 7 反应物1, 6a~7b过渡态的几何优化结构图
Figure 7 Optimized geometrical structures of transition states of reactants 1, 6a~7b
White: hydrogen, Gray: carbon, Blue: nitrogen, Purple: manganese, Red: oxygen, Orange: phosphorus
表 1 配合物1各反应态的NPA电荷
Table 1. NPA charge of reactant, transition state and product for complex 1
| RC | TS | PC | |
| Q-O-C6H4-NO2 | -0.665 | -0.692 | -0.968 |
| Q Corrole Mn | -0.527 | -0.308 | -0.198 |
 下载: 导出CSV
下载: 导出CSV
表 2 反应物1~5的部分键长
Table 2. Selected bond lengths of reactants 1~5
| Complex | Substituent | d(Mn-Oa)/nm | d(Mn-Ob)/nm | d(P-QC)/nm | d(P-Qa)/nm |
| 2 | -NH2 | 0.200 6 | 0.579 9 | 0.176 3 | 0.422 3 |
| 3 | -C2H5 | 0.197 6 | 0.586 6 | 0.177 6 | 0.420 8 |
| 4 | -OCH3 | 0.195 8 | 0.648 3 | 0.178 1 | 0.424 6 |
| 1 | -C6F5 | 0.195 7 | 0.569 0 | 0.176 2 | 0.420 3 |
| 5 | -CN | 0.193 6 | 0.576 4 | 0.176 5 | 0.419 7 |
下载: 导出CSV
表 3 反应物1~5过渡态的部分键长
Table 3. Selected bond lengths of transition states of reactants 1~5
| Complex | Substituent | d(Mn-Oa) / nm | d(Mn-Ob) / nm | d(P-Oc) / nm | d(P-Oa) / nm |
| 2 | -NH2 | 0.231 2 | 0.236 0 | 0.183 0 | 0.235 1 |
| 3 | -C2H5 | 0.225 5 | 0.231 4 | 0.184 0 | 0.228 3 |
| 4 | -OCH3 | 0.225 8 | 0.224 7 | 0.183 7 | 0.225 0 |
| 5 | -CN | 0.218 3 | 0.196 2 | 0.178 0 | 0.216 1 |
下载: 导出CSV
表 4 不同中位取代咔咯Mn(Ⅲ)配合物的E(2)
Table 4.
E(2) of Corrole Mn(Ⅲ) complexes with different meso substituents
| Complex | 2 | 3 | 4 | 1 | 5 |
| RC P-Oc | 1 143.06 | 1 104.57 | 1 082.79 | 1 242.30 | 1 195.27 |
| TS P-Oc | 1 045.30 | 1 072.09 | 1 066.86 | 1 115.02 | 1 145.11 |
| TS P-Oa | 194.58 | 228.44 | 250.09 | 286.96 | 304.14 |
下载: 导出CSV
表 5 不同中位取代咔咯Mn(Ⅲ)配合物的轨道能
Table 5.
Orbital energies of corrole Mn(Ⅲ) complexes with different meso substituents
| Complex | 2 | 3 | 4 | 1 | 5 |
| Substituent | -NH2 | -C2H5 | -OCH3 | -C6F5 | -CN |
| EHOMO | 0.052 2 | 0.028 7 | 0.018 8 | -0.003 9 | -0.020 3 |
| ELUMO | 0.070 9 | 0.077 8 | 0.081 9 | 0.069 2 | 0.059 0 |
下载: 导出CSV
表 6 不同取代基配合物水解反应的反应能垒
Table 6.
6 Reaction energy barriers of hydrolysis reaction of the complexes with different substituents
| Complex | 2 | 3 | 4 | l | 5 |
| Energy barrier | 104.81 | 92.09 | 81.49 | 77.23 | 63.76 |
下载: 导出CSV
表 7 反应物1, 6a~7b的部分键长
Table 7. Selected bond length parameters of reactants 1, 6a~7b
| Complex | d(Mn-Oa) / nm | d(Mn-Ob) / nm | d(P-Oc) / nm | d(P-Oa) / nm |
| 6a | 0.197 0 | 0.566 0 | 0.177 6 | 0.415 7 |
| 6b | 0.196 9 | 0.579 4 | 0.177 3 | 0.417 3 |
| 7a | 0.196 4 | 0.570 1 | 0.176 8 | 0.417 8 |
| 7b | 0.196 0 | 0.564 9 | 0.177 0 | 0.415 3 |
| l | 0.195 7 | 0.569 0 | 0.176 2 | 0.420 3 |
下载: 导出CSV
表 8 反应物1, 6a~7b过渡态的部分键长
Table 8. Selected bond length parameters of transition states of reactants 1, 6a~7b
| Complex | d(Mn-Oa) / nm | d(Mn-Ob) / nm | d(P-Oc) / nm | d(P-Oa) / nm |
| 6a | 0.226 7 | 0.223 3 | 0.183 5 | 0.224 9 |
| 6b | 0.228 9 | 0.220 7 | 0.183 1 | 0.222 6 |
| 7a | 0.230 1 | 0.2156 | 0.182 3 | 0.221 2 |
| 7b | 0.226 4 | 0.217 3 | 0.182 1 | 0.224 7 |
| 1 | 0.229 0 | 0.209 6 | 0.181 1 | 0.220 6 |
下载: 导出CSV
表 9 不同中位取代咔咯Mn(Ⅲ)配合物的二级微扰稳定化能
Table 9.
E(2) of corrole Mn(Ⅲ) complexes with different meso substituents
| Complex | 6a | 6b | 7a | 7b | 1 |
| RC P-Oc | 1 102.52 | 1 106.36 | 1 126.00 | 1 122.75 | 1 242.30 |
| TS P-Oc | 1 076.22 | 1 084.30 | 1 101.43 | 1 042.12 | 1 115.02 |
| TS P-Oa | 250.38 | 264.43 | 281.23 | 247.54 | 286.96 |
下载: 导出CSV
表 10 不同中位取代咔咯Mn(Ⅲ)配合物的轨道能
Table 10.
Orbital energies of corrole Mn(Ⅲ) complexes with different meso substituents
| Complex | 6a | 6b | 7a | 7b | 1 |
| EH0M0 | 0.015 2 | 0.013 9 | 0.003 7 | 0.004 4 | -0.003 9 |
| ELUM0 | 0.076 6 | 0.078 1 | 0.074 1 | 0.074 2 | 0.069 2 |
下载: 导出CSV
表 11 不同取代基配合物水解反应的反应能垒
Table 11.
11 Reaction energy barriers of hydrolysis reaction of the complexes with different substituents
| Complex | 6a | 6b | 7a | 7b | 1 |
| Energy barrier | 85.74 | 85.56 | 82.26 | 82.04 | 77.23 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
