0.
引言
氨(NH3 )不仅是人造氮肥的重要氮源,而且也是重要的储氢载体[1 2 。大气中氮气含量接近80%,因此合成氨的氮源非常丰富。众所周知,氮分子十分稳定,其中N≡N键的键能高达940 kJ·mol-1 ,N2 很难被活化。目前,工业合成氨仍然延用传统的哈伯合成氨法,但该合成氨技术需要高温高压的条件,并且其制氢及合成氨工艺中均会产生大量CO2 气体[3 4 。因此,寻找廉价、高效、绿色节能的合成氨技术具有重要意义。将合成氨反应的氢源由H2 换为H2 O,在常温常压的反应条件下与N2 直接反应生成NH3 是合成氨新途径。迄今为止,已经出现了生物固氮、电催化、光电催化以及光催化等研究方向[5 14 。尤其是以太阳光为驱动力的光催化合成氨作为可持续的绿色工艺引起了人们广泛关注[15 16 。最近,已经出现了以TiO2 、ZnO、BiOBr及g-C3 N4 为代表的光催化剂被应用于光催化合成氨[17 19 ,但光催化合成氨效率仍然较低。ZnO是一种廉价的、含较宽直接带隙(3.37 eV)的氧化物半导体材料[20 ,具有受激发电子可以稳定存在,电子迁移率高等特点,广泛应用于光催化领域[21 。但由于ZnO光生电子与空穴的复合率高且有光腐蚀现象,使其存在稳定性较低,容易被光生空穴氧化分解等缺陷[21 22 。研究发现,通过复合等手段能有效地提高其稳定性[23 。Janet等通过在ZnO上负载Pt来降低其电子与空穴的复合,但其光催化合成氨性能并不理想[24 。理论和实验研究表明,空位(如氧空位及氮空位等)可以有效地捕获激发电子,并将其转移到吸附氮分子的反键轨道(π *),从而有效地活化氮分子[25 26 。催化剂制备过程中容易同时制造体相氧空位和表面氧空位,由于体相氧空位更加有利于电子-空穴的复合导致催化效率降低,而催化剂表面氧空位比体相氧空位更有利于活化氮分子[27 28 。因此,通过改性或修饰等方法限制催化剂体相氧空位形成,尽量暴露表面氧空位对活化氮分子具有重要意义且具有一定的挑战性。近期研究发现,引入Fe3+ 可以抑制光生电子-空穴对的复合,从而提高光催化活性,由于Fe3+ 位点上空的d轨道不仅可以作为吸附和活化N2 分子的活性位点,还可以促进N2 分子与催化剂之间的电荷转移,从而显著提高氮的光固化能力[29 。因此,研究Fe2 O3 与ZnO的复合光催化剂对提高光催化合成氨具有一定意义。
我们以乙二醇作为还原剂,采用溶剂热法制备合成了富表面氧空位的Fe2 O3 /ZnO催化剂,利用X射线衍射(XRD)、透射电镜(TEM)、电子顺磁共振(EPR)、紫外-可见漫反射(UV-Vis DRS)、荧光光谱(PL)及光电流(PC)对Fe2 O3 /ZnO催化剂进行表征,并考察了Fe2 O3 /ZnO催化剂在常温、常压及无牺牲剂下的光催化合成氨的性能。通过引入氧化铁纳米粒子,不仅有效地控制了体相氧空位浓度,而且其丰富的表面氧空位及有效的电子-空穴分离能显著地提高催化剂的催化性能,为设计高效稳定的合成氨光催化剂提供了新的研究思路。
1.
实验部分
1.1
试剂与仪器
无水乙醇购于安徽安特食品股份有限公司;二水合乙酸锌(分析纯),氢氧化钠(分析纯),四水合酒石酸钾钠(分析纯),碘化钾(分析纯)购于西陇科学股份有限公司;氯化铁(化学纯)购于国药集团化学试剂有限公司;N2 (99.999%)购于杭州今工特种气体有限公司。
XRD是在Dutch PANalytical公司的X′ Pert PRO X射线衍射仪上表征的,使用Cu Kα 作为辐射源(λ =0.154 06 nm),电压、电流、扫描速率和扫描范围分别为40 kV、40 mA、10°·min-1 和10°~80°;SEM使用FEI的Nano nova 450型场发射扫描电子显微镜来表征;TEM使用FEI公司的Tecnai F30型场发射透射电子显微镜来表征;XPS是在Thermo Fischer ESCALAB 250Xi电子能谱仪上表征的,以Al Kα 作为X射线激发源,电子能量为1 486.6 eV,电子结合能通过C1s 峰(284.80 eV)来校准;UV-Vis DRS使用的是Shimadzu UV2550型号的紫外-可见光谱仪,将BaSO4 作为参比样对材料进行表征;PL是在室温下使用JY FluoroLog-3型号的荧光光谱仪来进行表征的,使用氙灯作为激发光源,激发波长为330 nm;EPR是由Bruker a300在室温下表征的,调制频率为100 kHz,中心磁场为0.342 T,扫场宽度为0.6 T。光电流测试是在标准的三电极系统在CHI 760E工作站上进行的,光源为300 W氙灯,以催化改性的导电玻璃(FTO,1 cm×2 cm)为工作电极,饱和甘汞电极(SCE)和铂电极作为参比电极和对电极,电解液为pH=6.84的磷酸溶液。其中工作电极的制备如下:将100 mg催化剂超声分散在含1 mL无水乙醇的混合溶液中,然后将200 μL制备好的混合溶液滴在导电玻璃上,在室温下干燥,最后将导电玻璃在30 ℃的真空干燥箱中干燥24 h。
1.2
实验过程
1.2.1
催化剂制备过程
Fe2 O3 制备:称取0.9 g FeCl3 溶解到50 mL蒸馏水中,逐滴加入15 mL NaOH溶液(1 mol·L-1 ),搅拌30 min后,将混合溶液转移至100 mL水热反应釜中,密封,在200 ℃下反应10 h。反应结束后,冷却至室温,离心10 min,转速为5 000 r·min-1 ,用去离子水和无水乙醇各洗涤2次,所得样品在60 ℃干燥12 h后在500 ℃下煅烧3 h,经研磨得Fe2 O3 粉末。
Fe2 O3 /ZnO催化剂制备:分别称取5、10、15、20 mg的Fe2 O3 粉末,分散在40 mL乙二醇中,并以700 r·min-1 的转速搅拌30 min,逐滴加入7.5 mL的Zn(CH3 COO)2 溶液(0.2 mol·L-1 ),继续搅拌30 min,缓慢逐滴加入2.5 mL NaOH溶液(3.6 mol·L-1 ),保持转速搅拌2 h。将混合溶液转移至100 mL水热反应釜中,在160 ℃下反应12 h。反应结束后,冷却至室温,5000 r·min-1 下离心10 min,用去离子水和无水乙醇各洗涤2次,将所得样品在60 ℃干燥12 h,研磨,将制得催化剂按Fe2 O3 的物质的量分数分别记为2%Fe2 O3 /ZnO、4%Fe2 O3 /ZnO、6%Fe2 O3 /ZnO和8%Fe2 O3 /ZnO。
1.2.2
光催化实验过程
称取10 mg上述制备好的催化剂超声分散在40 mL超纯水中,超声分散完全后,将分散液转移至反应釜中,密封,黑暗避光通30 min N2 ,使釜内处于鼓泡状态,使反应液与氮气达到吸附平衡,同时也为了排尽釜内氧气。30 min之后,打开氙灯以模拟太阳光进行光催化反应,氙灯功率为300 W。光催化反应结束后,将反应液转移到50 mL离心管,5 000 r·min-1 下离心10 min,取离心后的上层清液用0.22 μm的滤膜再过滤至25 mL比色管中,定容,采用纳氏法定量检测氨浓度。光催化合成氨的重复性实验是将进行了1 h光催化实验的反应液,进行高速离心分离,收集固体进行60 ℃干燥12 h,再次进行光催化合成氨实验,以此重复实验5次。
2.
结果与讨论
2.1
X射线粉末衍射表征
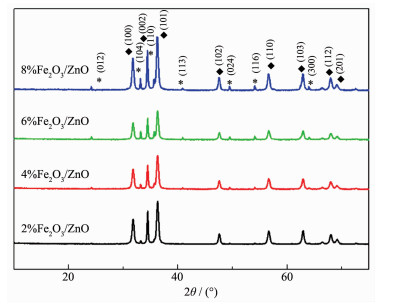
Fe2 O3 /ZnO纳米复合材料结构通过XRD表征,结果如图 1 2 O3 /ZnO的衍射峰角度没有偏移,表明Fe2 O3 与ZnO的复合没有影响ZnO的晶体结构。另外,在24.16°、33.19°、35.63°、40.93°、49.55°、54.18°、64.11°处对应Fe2 O3 衍射峰(PDF No.33-0664),分别对应(012)、(104)、(110)、(113)、(024)、(116)、(300)晶面。
图 1
2.2
SEM-mapping和TEM表征
图 2 2 O3 /ZnO纳米复合材料SEM及mapping图,黄色,红色和紫色点分别代表Zn,O和Fe元素。由图可知所有元素均匀分布在所制备的光催化剂中,这表明Fe2 O3 与ZnO是均匀分布的。
图 2
图 3 2 O3 /ZnO样品的TEM和HRTEM图,进一步表征了Fe2 O3 /ZnO结构。由图 3a 2 O3 /ZnO纳米复合材料是由ZnO纳米短棒和Fe2 O3 六方形纳米颗粒组成的,并且ZnO纳米短棒与Fe2 O3 纳米颗粒之间形成明显的异质界面(图 3b 图 3c d 2 O3 纳米结构的HRTEM图,分别对应ZnO暴露的(102)晶面和Fe2 O3 暴露的(104)晶面;通过FFT转换(内插图),可以清晰地看到Fe2 O3 纳米颗粒为单晶结构,其中ZnO的晶格间距约为0.191 nm,Fe2 O3 的晶面间距为0.273 nm。
图 3
2.3
XPS表征
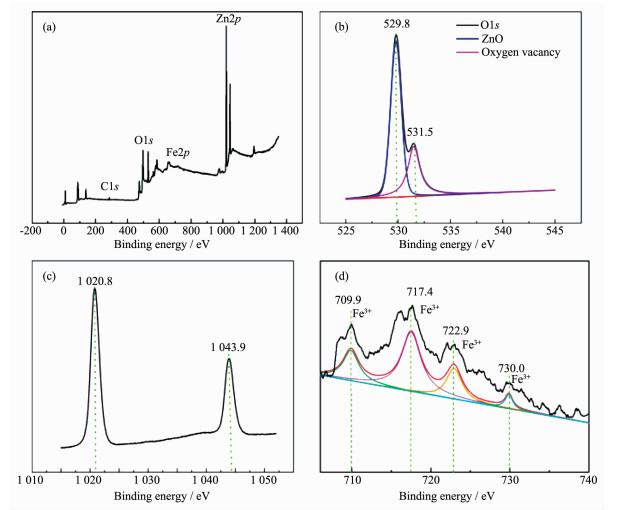
为了研究催化剂的组成及价态,对4%Fe2 O3 /ZnO样品进行了XPS表征。图 4a 2 O3 /ZnO样品的全谱图,由图可以看出存在Zn、Fe、O和C元素,其中C元素来自仪器,用于校正图谱数据。图 4b s 的图谱,位于529.8和531.5 eV处拟合到2个对称峰,表明样品中存在2种O物种,分别对应ZnO的晶格氧和表面吸附氧(对应氧空位)[30 。图 4c p 3/2 和Zn2p 1/2 ,表明ZnO的Zn2+ 状态。图 4d p 的图谱,717.4及722.9 eV分别对应Fe2p 3/2 和Fe2p 1/2 ,717.4和730.0 eV对应于Fe3+ 的卫星峰,表明在催化剂Fe2 O3 /ZnO中铁以Fe3+ 形式存在[31 。
图 4
2.4
光催化合成氨性能
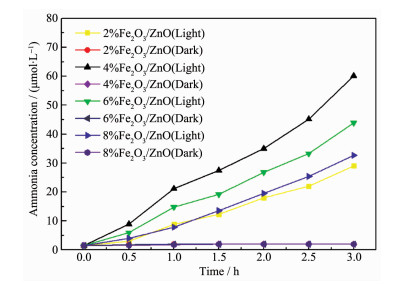
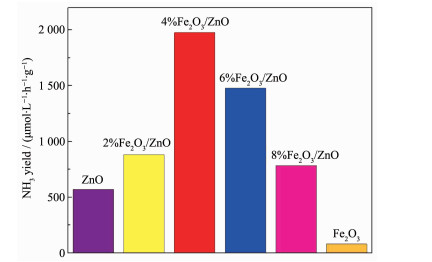
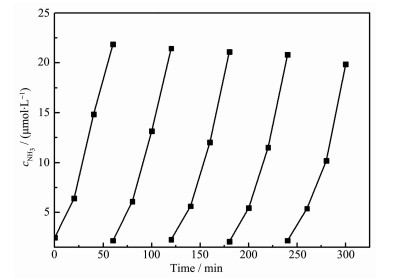
图 5 2 O3 /ZnO在模拟太阳光下和黑暗条件下生成氨的浓度随时间变化的曲线。黑暗条件下,复合催化剂均没有光催化效果。在光照下,生成氨的浓度随时间增加而增加,其中,以4%Fe2 O3 /ZnO作为光催化的反应体系生成氨的浓度最高,光催化3 h氨浓度达到60 μmol·L-1 。进一步研究不同Fe2 O3 含量的催化剂对光催化合成氨效率的影响,结果如图 6 2 O3 含量的增加,光催化合成氨的效率增加,当Fe2 O3 含量达到4%时,光催化剂有较高的合成氨反应效率,其合成氨效率达到2 059 μmol·L-1 ·g-1 ·h-1 (催化反应体系为40 mL,也可换算为82.4 μmol·g-1 ·h-1 ),远高于纯ZnO(600 μmol·L-1 ·g-1 ·h-1 )和Fe2 O3 (120 μmol·L-1 ·g-1 ·h-1 )光催化剂的光催化效率。进一步增加Fe2 O3 含量,光催化合成氨效率反而随之下降。保持实验条件不变,4%Fe2 O3 /ZnO催化剂进行多次回收循环实验(图 7 -1 ,催化剂的稳定性较好。与其他已经报道的光催化合成氨数据对比(表 1 2 O3 /ZnO显示较好的光催化合成氨效率。
图 5
图 6
图 7
表 1
2.5
EPR表征
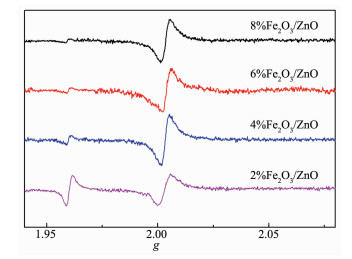
催化剂的氧空位种类及氧空位的浓度是光催化合成氨的重要因素,因此,进一步对Fe2 O3 /ZnO样品进行了EPR表征。图 8 2 O3 /ZnO样品的EPR图谱,Fe2 O3 /ZnO样品在g =1.96处的有明显信号,说明捕获到了来自体相氧空位的不成对电子[39 ;在g =2.003处观察到较强的顺磁对称信号,是由于捕获到了在表面氧空位和其他表面缺陷位点的未成对电子。当Fe2 O3 含量为2%时,在g =1.96和2.003处均出现了顺磁信号,表明乙二醇可以在水热过程中在Fe2 O3 /ZnO制造出丰富的体相氧空位和表面氧空位。随着Fe2 O3 含量的增加,g =1.96的信号减弱,当Fe2 O3 含量增至4%时,g =2.003处的信号增强,说明ZnO在生长过程中与Fe2 O3 纳米粒子的复合,形成了异质界面,从而导致体相氧空位的浓度逐渐减小,表面氧空位的浓度逐渐增大;随着Fe2 O3 含量增至6%和8%时,g =2.003处的信号较4%含量Fe2 O3 的样品降低,说明进一步增加含量,Fe2 O3 覆盖了表面氧空位,从而导致表面氧空位浓度降低,该表征结果与光催化合成氨效率规律一致。根据前文合成氨数据可知,4%Fe2 O3 /ZnO的光催化合成氨效率最大,表明催化剂表面氧空位是光催化合成氨的重要活性位点[40 。
图 8
2.6
UV-Vis DRS表征
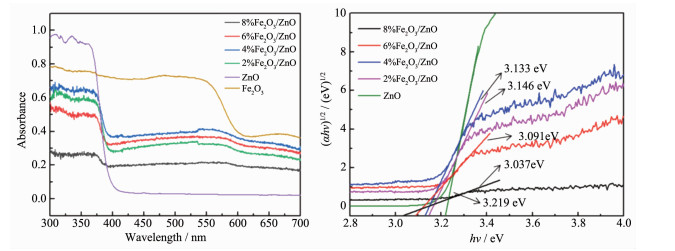
众所周知,纯ZnO禁带宽度为3.37 eV,在UV光下吸收能力强,但是在可见光区范围内对光几乎没有响应。通过控制Fe2 O3 含量可以调控ZnO对紫外-可见光的吸收区域。图 9 2 O3 及Fe2 O3 /ZnO样品的UV-Vis DRS及禁带宽度图谱。由图 9a 2 O3 的含量直接影响了ZnO对紫外及可见光区的吸收,在540~600 nm范围内出现了宽吸收峰,表明Fe2 O3 与ZnO的复合能够有效的增大催化剂对可见光范围的吸收,当Fe2 O3 含量达到4%时,对紫外区域光的吸收最高,这可能是4%Fe2 O3 /ZnO光催化效率较高的主要原因之一。图 9b 2 O3 /ZnO纳米复合材料的禁带宽度示意图,单纯的ZnO的禁带宽度为3.219 eV,Fe2 O3 的掺杂使ZnO纳米复合材料的禁带宽度逐渐降低,且随着Fe2 O3 掺杂量的增大,光催化剂的禁带宽度从3.219 eV一直降低到3.037 eV,禁带宽度的降低,在光照下有利于电子从价带往导带跃迁。
图 9
2.7
PL光谱表征
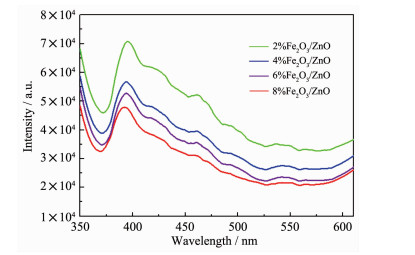
为了进一步研究Fe2 O3 /ZnO纳米复合材料中光生电子空穴的分离与复合效率,进行了PL光谱表征。图 10 2 O3 /ZnO纳米复合材料的PL光谱(激发波长为330 nm)。在可见光区(400~500 nm)产生的荧光是由于光生电子-空穴复合所致[41 。一般说来,电子-空穴越容易复合,荧光强度越强;反之,电子-空穴分离效率较高,荧光强度越弱[42 。由于Fe2 O3 纳米粒子与ZnO产生强相互作用,从而形成了异质界面,有利于光电子-空穴的分离,且随着Fe2 O3 含量的增加,Fe2 O3 /ZnO复合物的荧光强度明显下降,表明Fe2 O3 的引入能够有效地抑制电子与空穴的复合,这也是提高光催化活性主要因素。
图 10
2.8
光电流表征
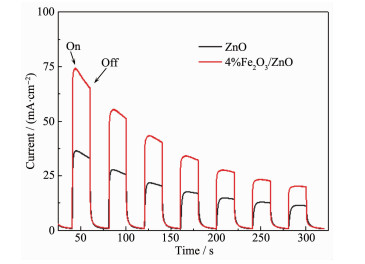
图 11 2 O3 /ZnO与纯ZnO的光电流响应对比图。与纯ZnO相比,在相同光源的照射下,4%Fe2 O3 /ZnO光电流明显增大。这是由于ZnO与Fe2 O3 形成的异质界面可以迅速地将光电子从ZnO导带(CB)转移到Fe2 O3 导带(CB),从而有效地抑制光生空穴与电子的复合,显著提高了光生电子-空穴的分离效率。
图 11
2.9
催化剂反应后XRD表征
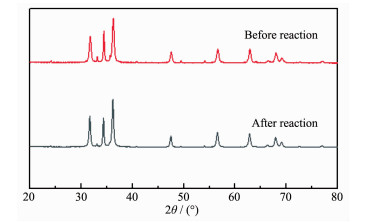
为了进一步研究光催化剂的稳定性,对4%Fe2 O3 /ZnO光催化反应前后的XRD图进行了对比(图 12 2 O3 /ZnO光催剂稳定性很好。
图 12
2.10
光催化反应机理
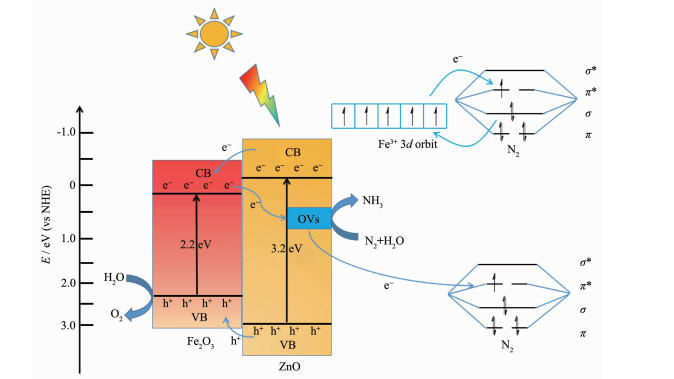
光催化合成氨的机理如图 13 2 O3 形成异质界面,ZnO导带(CB)上光生电子可以转移到Fe2 O3 导带(CB)上,再结合Fe2 O3 自身产生的光电子,转移到ZnO表面氧空位上。当氮分子吸附在催化剂表面时,表面氧空位作为诱导缺陷态,将电子转移到吸附的N2 分子的反键轨道(π *)上,从而降低N≡N的键能;另外,由于Fe3+ 的d 轨道上含有半充满电子,很容易提供或接受电子给其他物质,Fe3+ 将其d 轨道上不成对的电子提供给N2 分子的反键轨道(π *),同时N2 的σ 轨道上的电子转移到Fe3+ 的d 轨道上,由此活化N2 ,从而与表面氧空位起到协同活化单分子的作用。活化的氮分子(·N2 )与H+ 反应生成各种中间一系列过渡态分子(N2 H、N2 H2 、N2 H4 、N2 H5 + ),最终反应生成NH3 [43 44 。ZnO表面引入Fe2 O3 纳米粒子后,有效地降低了催化剂的体相氧空位浓度,并且显著提高了光催化剂在可见光区域的光吸收;另外,Fe2 O3 和ZnO形成表面异质结,提高了光生电子与空穴的分离效率,避免了电子-空穴复合,又进一步有利于光催化合成氨的进行。
图 13
3.
结论
通过溶剂热法将ZnO与Fe2 O3 复合,制备出一种富有表面氧空位的Fe2 O3 /ZnO光催化剂。将其应用在光催化合成氨反应中,能够提高TiO2 的光催化活性。当Fe2 O3 的复合量达到4%时,催化活性最高,NH3 的产率达到2 059 μmol·L-1 ·g-1 ·h-1 。表征的结果表明氧空位与过渡金属Fe协同作用于吸附活化N2 ,同时Fe2 O3 的复合降低了体相氧空位的浓度,抑制了光生电子与空穴的复合,提高了光催化合成氨性能。这一结果为制备其他高效且稳定的复合催化剂体系提供一种新的思路。

 下载:
下载:



 下载:
下载:














