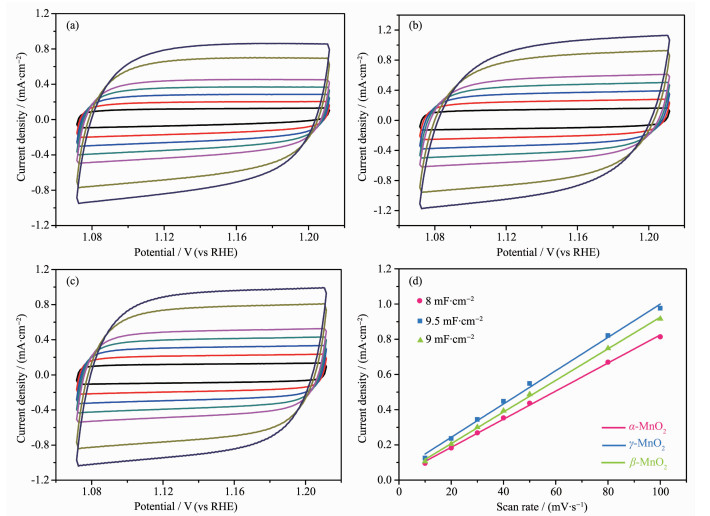
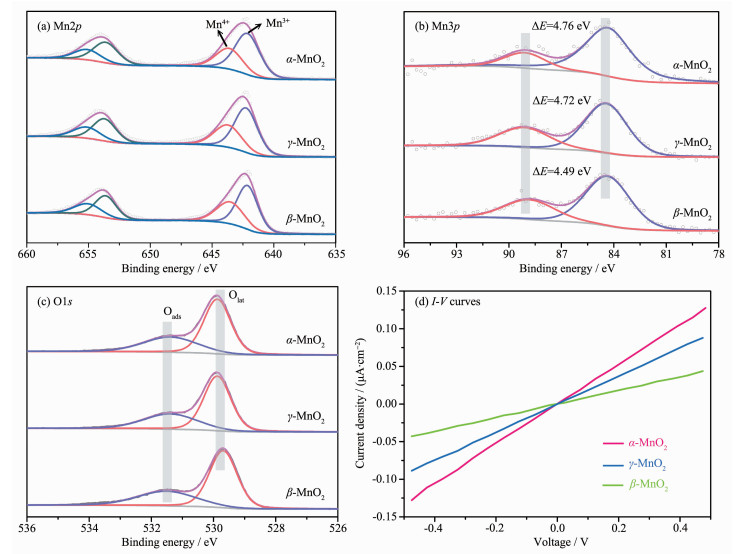
-
[1]
Jin H Y, Guo C X, Liu X, et al. Chem. Rev., 2018, 118:6337-6408 doi: 10.1021/acs.chemrev.7b00689
-
[2]
Zhang W, Lai W Z, Cao R. Chem. Rev., 2017, 117:3717-3797 doi: 10.1021/acs.chemrev.6b00299
-
[3]
Hosseini-Benhangi P, Kung C H, Alfantazi A, et al. ACS Appl. Mater. Interfaces, 2017, 9:26771-26785 doi: 10.1021/acsami.7b05501
-
[4]
Xia W, Mahmood A, Liang Z B, et al. Angew. Chem. Int. Ed., 2016, 55:2650-2676 doi: 10.1002/anie.201504830
-
[5]
Ge X M, Sumboja A, Wuu D, et al. ACS Catal., 2015, 5:4643-4667 doi: 10.1021/acscatal.5b00524
-
[6]
Hu C G, Dai L M. Angew. Chem. Int. Ed., 2016, 55:11736-11758 doi: 10.1002/anie.201509982
-
[7]
Lv L, Zha D C, Ruan Y J, et al. ACS Nano, 2018, 12:3042-3051 doi: 10.1021/acsnano.8b01056
-
[8]
Yan X C, Jia Y, Chen J, et al. Adv. Mater., 2016, 28:8771-8778 doi: 10.1002/adma.201601651
-
[9]
冯艳, 吴剑波, 张晓玲, 等.无机化学学报, 2019, 35(4):569-579 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20190401&journal_id=wjhxxbcnFENG Yan, WU Jian-Bo, ZHANG Xiao-Ling, et al. Chinese J. Inorg. Chem., 2019, 35(4):569-579 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20190401&journal_id=wjhxxbcn
-
[10]
Osgood H, Devaguptapu S V, Xu H, et al. Nano Today, 2016, 11:601-625 doi: 10.1016/j.nantod.2016.09.001
-
[11]
Wang Y, Li J, Wei Z D. J. Mater. Chem. A, 2018, 6:8194-8209 doi: 10.1039/C8TA01321G
-
[12]
Mosa I M, Biswas S, El-Sawy A M, et al. J. Mater. Chem. A, 2016, 4:620-631 doi: 10.1039/C5TA07878D
-
[13]
Liu B, Sun Y L, Liu L, et al. Adv. Funct. Mater., 2018, 28:1704973 doi: 10.1002/adfm.201704973
-
[14]
Cheng F Y, Su Y, Liang J, et al. Chem. Mater., 2010, 22:898-905 doi: 10.1021/cm901698s
-
[15]
Cao Y L, Yang H X, Ai X P, et al. J. Electroanal. Chem., 2003, 557:127-134 doi: 10.1016/S0022-0728(03)00355-3
-
[16]
Zhang X H, Li B X, Liu C Y, et al. Mater. Res. Bull., 2013, 48:2696-2701 doi: 10.1016/j.materresbull.2013.03.025
-
[17]
Meng Y T, Song W Q, Huang H, et al. J. Am. Chem. Soc., 2014, 136:11452-11464 doi: 10.1021/ja505186m
-
[18]
Duan J J, Chen S, Dai S, et al. Adv. Funct. Mater., 2014, 24:2072-2078 doi: 10.1002/adfm.201302940
-
[19]
Cheng G, Yu L, He B B, et al. Appl. Surf. Sci., 2017, 409:223-231 doi: 10.1016/j.apsusc.2017.02.218
-
[20]
Liu S X, Zhang R, Lv W X, et al. Int. J. Electrochem. Sci., 2018, 13:3843-3854
-
[21]
Lu H B, Wang S M, Zhao L, et al. J. Mater. Chem., 2011, 21:4228-4234 doi: 10.1039/c0jm03390a
-
[22]
Zhong X, Sun Y Y, Chen X L, et al. Adv. Funct. Mater., 2016, 26:5778-5786 doi: 10.1002/adfm.201601732
-
[23]
Gao T, Fjellvag H, Norby P. Anal. Chim. Acta, 2009, 648:235-239 doi: 10.1016/j.aca.2009.06.059
-
[24]
Cheng S, Yang L F, Chen D C, et al. Nano Energy, 2014, 9:161-167 doi: 10.1016/j.nanoen.2014.07.008
-
[25]
Gao T, Glerup M, Krumeich F, et al. J. Phys. Chem. C, 2008, 112:13134-13140 doi: 10.1021/jp804924f
-
[26]
Julien C, Massot M, Rangan S, et al. J. Raman Spectrosc., 2002, 33:223-228 doi: 10.1002/jrs.838
-
[27]
Lei K X, Cong L, Fu X R, et al. Inorg. Chem. Front., 2016, 3:928-933 doi: 10.1039/C6QI00056H
-
[28]
Zhang T R, Cheng F Y, Du J, et al. Adv. Energy. Mater., 2015, 5:1400654 doi: 10.1002/aenm.201400654
-
[29]
Zhang T R, Ge X M, Zhang Z, et al. ChemCatChem, 2018, 10:422-429 doi: 10.1002/cctc.201701192
-
[30]
Gu L Z, Jiang L H, Jin J T, et al. Carbon, 2015, 82:572-578 doi: 10.1016/j.carbon.2014.11.010
-
[31]
Liu X, Du J, Li C, et al. J. Mater. Chem. A, 2015, 3:3425-3431 doi: 10.1039/C4TA05995F
-
[32]
Ma T Y, Zheng Y, Dai S, et al. J. Mater. Chem. A, 2014, 2:8676-8682 doi: 10.1039/C4TA01672F
-
[33]
Xiao J W, Kuang Q, Yang S H, et al. Sci. Rep., 2013, 3:2300 doi: 10.1038/srep02300
-
[34]
Wang J, Wu Z X, Han L L, et al. Chin. Chem. Lett., 2016, 27:597-601 doi: 10.1016/j.cclet.2016.03.011
-
[35]
Niu Y L, Huang X Q, Zhao L, et al. ACS Sustainable Chem. Eng, 2018, 6:3556-3564 doi: 10.1021/acssuschemeng.7b03888
-
[36]
Thommes M, Kaneko K, Neimark A V, et al. Pure Appl. Chem., 2015, 87:1051-1069 doi: 10.1515/pac-2014-1117
-
[37]
Song W Q, Ren Z, Chen S Y, et al. ACS Appl. Mater. Interfaces, 2016, 8:20802-20813 doi: 10.1021/acsami.6b06103
-
[38]
Lee S H, Nam G, Sun J, et al. Angew. Chem. Int. Ed., 2016, 55:8599-8604 doi: 10.1002/anie.201602851
-
[39]
Lei K X, Han X P, Hu Y X, et al. Chem. Commun., 2015, 51:11599-11602 doi: 10.1039/C5CC03155A
-
[40]
Tang X F, Li Y G, Chen J L, et al. Microporous Mesoporous Mater., 2007, 103:250-256 doi: 10.1016/j.micromeso.2007.02.008
-
[41]
Hou J T, Li Y Z, Liu L L, et al. J. Mater. Chem. A, 2013, 1:6736-6741 doi: 10.1039/c3ta11566f
-
[42]
Li H J, Qi G S, Tana, et al. Appl. Catal. B, 2011, 103:54-61 doi: 10.1016/j.apcatb.2011.01.008
-
[43]
Galakhov V, Falub M, Bartkowski S, et al. Phys. Rev. B, 2002, 65:113102 doi: 10.1103/PhysRevB.65.113102
-
[44]
Stoerzinger K A, Risch M, Han B H, et al. ACS Catal., 2015, 5:6021-6031 doi: 10.1021/acscatal.5b01444
-
[45]
Suntivich J, Gasteiger H A, Yabuuchi N, et al. Nat. Chem., 2011, 3:546-550 doi: 10.1038/nchem.1069
-
[46]
Zeng Y X, Lai Z Z, Han Y, et al. Adv. Mater., 2018, 30:1802396 doi: 10.1002/adma.201802396
-
[47]
Li L, Feng X H, Nie Y, et al. ACS Catal., 2015, 5:4825-4832 doi: 10.1021/acscatal.5b00320
-
[48]
Cheng F Y, Zhang T R, Zhang Y, et al. Angew. Chem. Int. Ed., 2013, 52:2474-2477 doi: 10.1002/anie.201208582

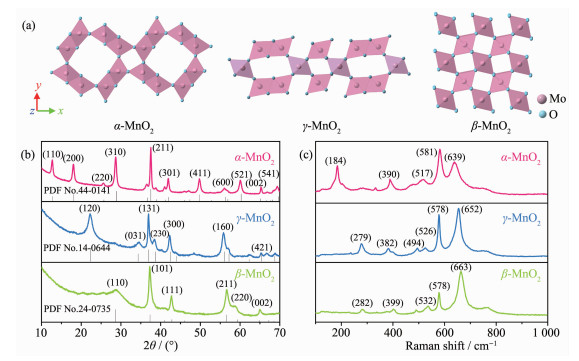
 下载:
下载:


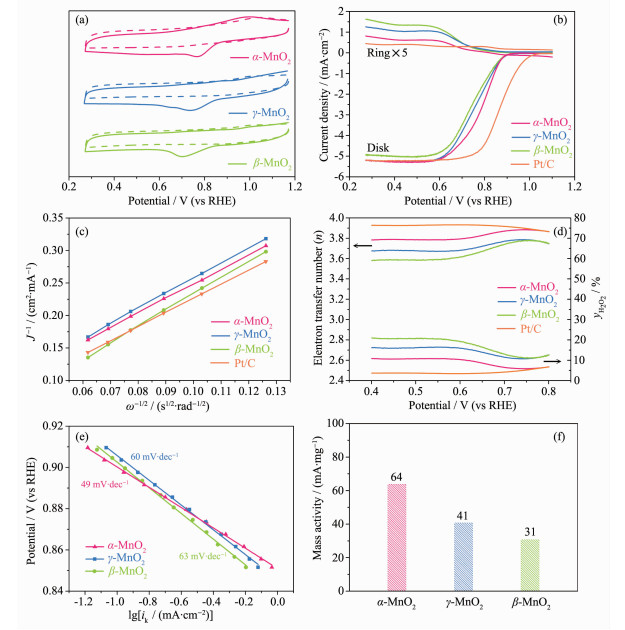
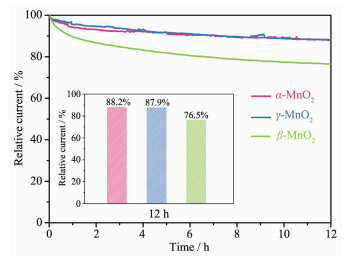
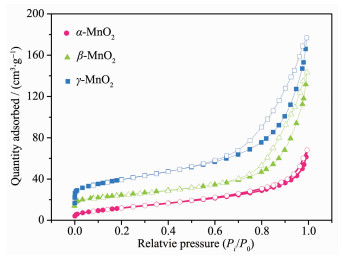

 下载:
下载:









