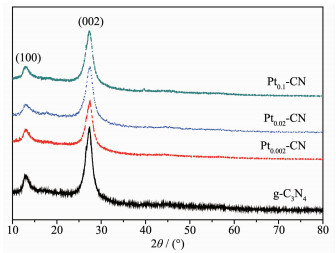
图 1.
g-C3N4和Pt2+/Pt0-CN样品的XRD图
Figure 1.
XRD patterns of g-C3N4 and Pt2+/Pt0-CN samples

有机染料和抗生素在自然界中很难降解,容易造成水体污染。例如,药品抗生素环丙沙星可以对人体产生肝脏和血液毒性,也可以使环境中的细菌对其产生抗性。所以环丙沙星在环境中会对生态系统和人类健康构成潜在威胁。半导体光催化氧化技术,可以利用太阳能将水体有机污染物分解矿化,具有绿色环保的优点。石墨相碳化氮(g-C3N4)是一种不含金属的n型半导体,具有无毒、高稳定、低成本的特点。它的带隙约为2.7 eV,具有良好的可见光吸收性能,因此在光催化领域引起了广泛的关注[1-2]。
设计独特的纳米结构和沉积贵金属是提高g-C3N4光催化活性的2种有效策略。具有独特纳米结构的g-C3N4往往具有更多的活性中心和高效的光捕获能力;在g-C3N4表面沉积贵金属纳米颗粒可以显著降低光生电子(e-)空穴(h+)对的复合几率[3-4]。例如,Lu等[5]用溶剂热法制备了高分散的Pt/g-C3N4纳米复合材料,当Pt含量为2%时,样品表现出最好的光催化性能,可见光下降解甲基橙和盐酸四环素分别为纯g-C3N4的7.82和4.30倍。Nagajyothi等[6]合成了银球形纳米粒子修饰的g-C3N4,发现在紫外光照射下降解孔雀石绿染料的光催化活性是g-C3N4的2.5倍。Zhang等[7]采用光沉积法制备了不同银含量的Ag/g-C3N4复合光催化剂,通过比较可见光下降解双氯芬酸的速率,发现Ag/g-C3N4光催化活性高于纯g-C3N4。Sun等[8]采用微波辅助多元醇法,合成了Ag/g-C3N4光催化剂并具有降解罗丹明B的优良可见光催化活性。
以上研究表明贵金属纳米粒子沉积是提高g-C3N4光催化性能的有效途径。但是报道的大部分贵金属纳米粒子是通过传统化学或光学还原方法直接沉积在g-C3N4块体的表面,因此,贵金属纳米粒子的电子捕获效应仅局限于g-C3N4的表面[9]。另外,报道中贵金属的使用含量普遍较高,制备成本高[10-11]。我们采用简单三聚氰胺和氯铂酸溶液共聚法制备了铂含量很低的Pt2+/Pt0-g-C3N4复合光催化剂,并探讨了掺杂微量铂对g-C3N4的组织结构、光吸收、光电性能和光催化性能的影响。
以三聚氰胺与氯铂酸溶液为原料,采用共缩聚法制备Pt2+/Pt0-CN。将1 g三聚氰胺和不同体积(0.01 mL、0.1 mL、0.5 mL)的浓度为4 g·L-1的氯铂酸水溶液加入到坩埚中,控制Pt与g-C3N4的理论质量之比分别为0.002%、0.02%、0.1%(样品分别记作Pt0.002-CN、Pt0.02-CN、Pt0.1-CN)。再加入10 mL水,搅拌均匀后超声分散10 min,接着置于烘箱中于100 ℃烘干水分,研磨后在520 ℃下煅烧2 h,升温速率为5 ℃·min-1。最后加无水乙醇对样品进行充分研磨并干燥,获得Pt2+/Pt0-CN样品。纯g-C3N4制备的方法一样,只是不加氯铂酸,其它步骤相同。
制备的光催化剂的物理化学性质通过一系列的表征手段进行分析。如X射线粉末衍射(XRD)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)、傅里叶变换红外光谱(FT-IR)、紫外可见漫反射光谱(UV-Vis DRS)、光电流测试、N2物理吸附-脱附和X射线光电子能谱(XPS)等。XRD测试采用铜靶(Cu Kα,λ=0.154 06 nm,工作电压为40 kV,电流为40 mA),扫描范围为10°~80°,速度10°·min-1。N2物理吸附测试条件为冷阱(液氮-195 ℃),脱气温度为120 ℃,脱气时间为1 h。SEM放大倍率5万~20万倍。TEM测试最大放大倍数为80万倍,点分辨率为0.24 nm。FT-IR测试以KBr为进行压片,压力为1.5 T,测试范围为500~4 000 cm-1。UV-Vis DRS测试以BaSO4作为参比,扫描范围200~800 nm。XPS测试:Al Kα1 486.6 eV。光电测试以氧化铟锡玻璃片制样,参比电极为饱和甘汞电极,工作电极为样品,辅助电极为Pt电极,电解液为0.1 mol·L-1 Na2SO4溶液,光源为300 W氙灯加420 nm滤光片,滤去波长小于420 nm的光。
以10 mg·L-1的染料罗丹明B、酸性橙Ⅱ和甲基橙以及15 mg·L-1的染料亚甲基蓝为模拟目标污染物降解对象,分别利用紫外-可见分光光度计在其特征吸收波长(552、483、460和661 nm)处测定染料浓度,以400 W金卤灯为光源。以10 mg·L-1环丙沙星为抗生素的目标降解物,在271 nm的特征吸收波长处测试其光催化降解浓度,利用350 W氙灯作为光源。光催化反应器为定制的双层石英玻璃反应器,为保证反应处于恒温条件,用循环冷却水控制水温。分别取30 mg催化剂和50 mL模拟污染物的水溶液混合,用磁力搅拌器搅拌使之充分分散。将反应器置于黑暗处磁力搅拌90 min,使样品与染料分子达到物理吸附-脱附平衡。接着开灯进行光照实验,在光照条件下,间隔一定时间对反应混合液进行取样。将取得样品用离心机进行离心去除固体颗粒,取上清液,用紫外可见分光光度计(UV-6300)测试污染物的吸光度随时间的变化从而计算出染料浓度的变化。根据下面公式计算出目标污染物的降解率:D=(C0-Ct)/C0×100%=(A0-At)/A0×100%,其中C0和Ct分别是污染物在光照下初始和t时刻的浓度,A0与At分别为污染物在光照下初始和t时刻的吸光度。
图 1为合成样品的XRD图。纯g-C3N4在2θ=12.7°和27.4°处有2个衍射峰,分别对应(100)和(002)晶面。(100)晶面由构成平面的3-s-三嗪结构单元所形成,(002)晶面由π共轭平面的石墨层状堆积而形成[12]。在Pt2+/Pt0-CN样品中可以清晰地观察到其衍射峰和纯g-C3N4的基本一样,表明掺杂Pt2+/Pt0没有改变g-C3N4的结构。但是Pt2+/Pt0-CN的XRD峰强度相对于g-C3N4变弱了,说明其结晶度不如g-C3N4。通过放大(002)晶面,发现(002)衍射峰从27.4°→27.6°→27.5°→27.3°发生略微的变化,表明随着Pt2+/Pt0含量的增加(002)晶面的衍射角先增大而后减小,意味着g-C3N4层间的距离先减小后增大[13]。在12.7°处的(100)峰略有变弱,这可能是由于Pt2+/Pt0被填充在g-C3N4面内的原因[14]。在所有Pt2+/Pt0-CN样品,均未观察到Pt粒子的衍射峰,这主要由于其含量特别低的缘故。
表 1表明,Pt2+/Pt0-CN的比表面积较g-C3N4明显增大。其中,Pt0.02-CN的比表面积最大,为32 m2· g-1。比表面积增大,主要是因为制备过程中加入氯铂酸改变了g-C3N4的组织结构。图 2为g-C3N4和Pt2+/Pt0-CN样品的N2吸附-脱附等温线和孔径分布曲线,等温线呈现典型的Ⅳ曲线,孔径主要分布在10~40 nm之间,表明g-C3N4和Pt2+/Pt0-CN样品都是具有类似介孔(2~50 nm)结构的材料。同时所有样品在相对压力为0.5~0.995(P/P0)处存在一个归属于H3类的迟滞环[15]。
 下载:
导出CSV
下载:
导出CSV
| Sample | Specific surface area / (m2 · g-1) | Pore volume / (cm3·g-1) | Pore size / nm |
| g-C3N4 | 4 | 0.04 | 34 |
| Pt0.002-CN | 27 | 0.12 | 18 |
| Pt0.02-CN | 32 | 0.10 | 12 |
| Pt0.1-CN | 20 | 0.07 | 14 |
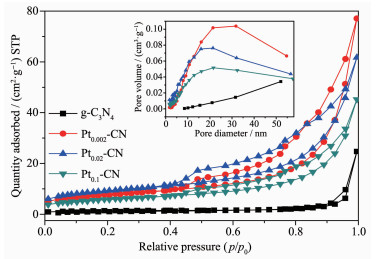
图 3展现了样品g-C3N4和Pt0.02-CN的形貌。图 3(a)是g-C3N4的扫描电镜图,从图中可以看出g-C3N4是由不规则的块状颗粒所组成,这是热聚合法合成的g-C3N4的典型结构特征。图 3(b)为Pt0.02-CN的扫描电镜图,可以看出在样品Pt0.02-CN内部存在一些孔洞。从图 3(c)与图 3(d)中可以清楚地看到,Pt0.02-CN中存在一些黑色的纳米粒子分散在g-C3N4的表面和内部,其平均粒径为5 nm。而在图 3(e)中,纯g-C3N4中没有那些黑点,结合XPS分析样品中存在Pt元素,分析这些黑点为Pt纳米粒子[9, 16-17]。另外,这些黑点大小不一、分布不均匀,这可能是由于三聚氰胺不溶于水,与氯铂酸溶液混合不均匀,导致煅烧时生成的Pt纳米粒子大小不均匀。

图 4(a)为g-C3N4和Pt2+/Pt0-CN的紫外-可见漫反射吸收光谱。纯g-C3N4在200~460 nm范围内表现出典型的吸收;随着Pt2+/Pt0含量的增加,光吸收逐渐增强,这主要是因为在样品中加入了Pt2+/Pt0物种,样品的颜色随着Pt2+/Pt0量的增加,逐渐变黑,从而降低了样品对光的反射[18]。此外,Pt2+/Pt0在测试波长范围内有比较强的光吸收,有可能来自于Pt2+/Pt0的优良的内在吸收[19]。由于g-C3N4是直接带隙半导体,因此,用(αhν)2=B(hν-Eg)方程计算制备光催化剂的带隙能,其中α、hν、Eg和B分别是吸收系数、光子能量、带隙和常数[20-21]。在图 4(b)中,带隙能根据上述(αhν)2与hν曲线的切线截距得到,其中g-C3N4、Pt0.002-CN、Pt0.02-CN、Pt0.1-CN的带隙能分别为2.70、2.72、2.76、2.73 eV。带隙能的增大可以增强光吸收能力,提高电子和空穴的氧化-还原能力,进而增强g-C3N4的光催化活性[22]。
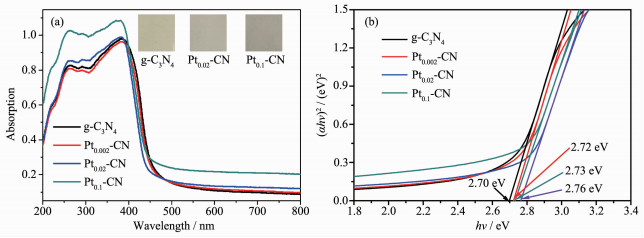
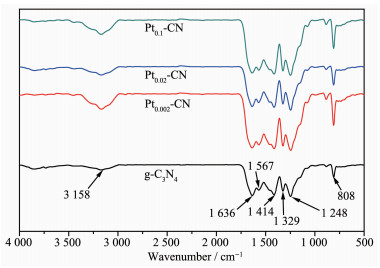
图 5为g-C3N4和Pt2+/Pt0-CN的红外光谱图。Pt2+/Pt0-CN样品的红外光谱吸收峰与g-C3N4类似。样品在808 cm-1处的吸收峰可归为3-s-三嗪环结构中碳氮环的弯曲振动特征峰[23]。在1 200~1 700 cm-1区域中的一系列吸收峰归于g-C3N4中碳氮杂环的伸缩振动峰[24]。在1 248 cm-1处的峰值与C-N(-C)-C或C-NH-C的伸缩振动相似,在1 329和1 636 cm-1处出现的振动峰是由杂环中的C-N和C=N振动吸收引起的,而在1 414 cm-1处的峰值是C=C的振动吸收。位于3 000~3 600 cm-1范围内的宽峰是胺基或OH基团的振动吸收峰[25]。值得注意的是,对于Pt2+/Pt0-CN,在1 200~1 700 cm-1范围内的一系列峰略有增强,这可能是由三嗪基元在纳米片层中排列更有序所致[26]。
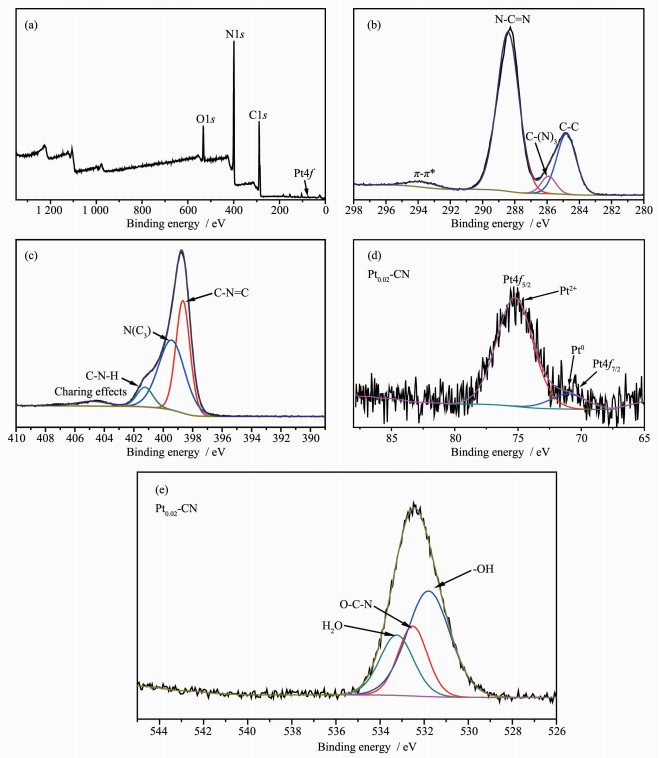
用XPS对Pt0.02-CN的表面元素组成和元素的化学状态进行分析。图 6(a)表明,样品由C、N和Pt元素组成,从而证实了复合材料中g-C3N4和Pt的存在。在图 6(a)中C1s谱可分为4个不同的峰。以284.8 eV为中心的峰通常归为表面上的C-C和C=C的碳[27];在285.91 eV处的峰属于sp3杂化碳(C-(N)3), 而在288.42 eV处的峰归属于芳香环(N=C-(N)2)中与N键合的sp2杂化碳原子[28];293.76 eV处的弱峰是由π-π*半线结构所引起[19],证明合成了g-C3N4。图 6(c)表明,N1s的结合能可分裂成398.66、399.43、401.24和404.80 eV四个峰,这些峰分别与sp2杂化的芳香氮原子(C=N-C),叔氮基((C)3-N),游离氨基((C)2 -NH,C-NH2)和正电荷在杂环中的局域化有关[22]。图 6(d) Pt4f峰可分为71.03 eV和75.24 eV两个峰。71.03 eV处的峰归为金属Pt0,75.24 eV处的峰归为Pt2+ [29]。因此, Pt在Pt0.02-CN样品中的价态为Pt2+和Pt0, 根据XPS分析,Pt2+和Pt0的质量百分比分别约为87.89%和12.11%。图 6(e)中O1s可分为3个峰,分别对应于羟基(-OH,531.80 eV)、O-C-N(532.52 eV)和化学吸附的H2O(533.24 eV),样品中所含的氧(O-C-N)可能来自于加热处理过程中空气中的氧[22]。
利用荧光光谱分析了电子和空穴的分离和复合情况[30]。从图 7(a)中可以看出,在室温下,当激发波长为365 nm时,g-C3N4在400~600 nm范围内表现出较强的发射峰,其中心位于450 nm左右。很明显Pt2+/Pt0-CN的光致发光强度较g-C3N4降低,表明Pt2+/Pt0存在使样品的光生电子和空穴的复合几率变小,提升了光生载流子的分离效率[31-32]。随着Pt2+/Pt0含量的增加,复合材料的PL强度一直呈现降低的趋势,这由于负载一定范围内的Pt2+/Pt0有利于捕获光生电子(e-),从而促进光生电子和空穴的分离。
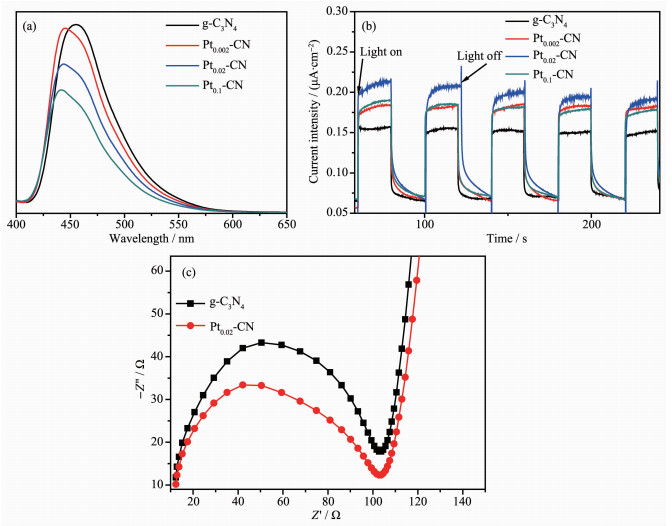
进一步利用可见光(λ > 420 nm)照射下的光电流响应评价催化剂的光生电子和空穴的分离效率[33]。由图 7(b)可见,当可见光被交替打开和关闭时,催化剂的光电流响应可再现地切换, 表明Pt2+/Pt0-CN样品具有较高的光电稳定性。光电流强度的增加表明电子和空穴的复合率较低。其中,Pt2+/Pt0可充当电子库,捕捉从激发的g-C3N4中迁移的电子,这将促进光生电子和空穴的分离。但是Pt2+/Pt0负载量过高时,对催化剂的光电性能造成不利的影响。与Pt0.02-CN相比,Pt0.1-CN的光电流响应有所下降。在Pt2+/Pt0掺杂的g-C3N4,当光源打开时,光电流响应值逐渐增加到一个恒定值,当光源关闭时,光电流逐渐减小到零。g-C3N4和复合物的光电流曲线形状存在较大的差异,这可能是因为当Pt2+/Pt0掺杂到g-C3N4时,g-C3N4导带上的光生电子转移到Pt2+/Pt0上。因此,光电流是由Pt2+/Pt0上存储的电子产生的,而不是直接从g-C3N4导带转移过来的电子,导致光电流逐渐增大。另一方面,当光源被关闭时,由于Pt2+/Pt0的电子储存作用,电子逐渐从Pt2+/Pt0上释放出来,并进一步转移到工作电极上,导致光电流逐渐减小到零[34]。
图 7(c)为Pt0.02-CN和g-C3N4的电化学阻抗谱(EIS)测试结果。电化学阻抗谱表明当电子和空穴的分离效率越高,电极表面的电荷转移电阻就越小,在电化学阻抗谱图上显示出较小的弧半径[13, 35]。Pt0.02-CN对应的弧半径小于g-C3N4的弧半径,与光催化性能测试一致。
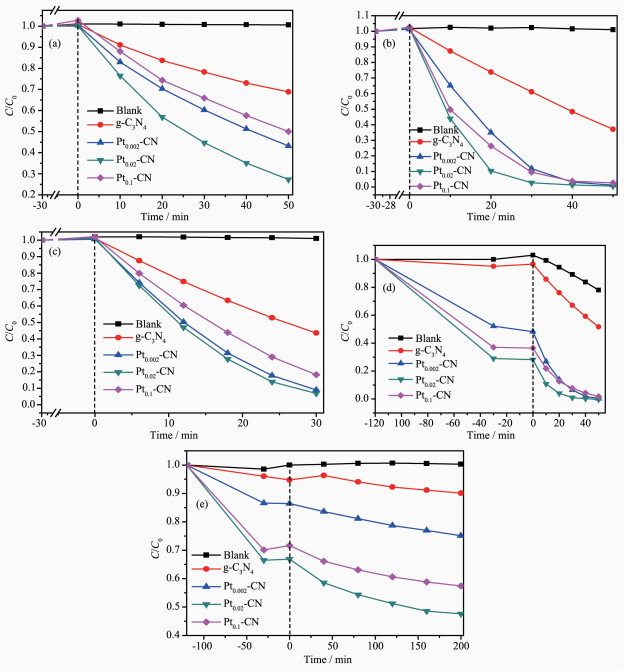
以400 W金卤灯为光源,以罗丹明B、酸性橙Ⅱ、亚甲基蓝、甲基橙为模拟目标污染物作为降解对象,对光催化剂的光催化性能进行评价。从图 8可以明显看出Pt2+/Pt0-CN复合材料光催化活性明显优于g-C3N4。相对于g-C3N4,Pt0.02-CN对罗丹明B、酸性橙Ⅱ、亚甲基蓝和甲基橙的降解性能分别提升了64%、39%、52%和42%。用350 W氙灯为光源,以环丙沙星为目标降解物,发现相对于g-C3N4,Pt0.02-CN的降解性能提升了43%。Pt2+/Pt0-CN复合材料光催化剂性能明显增强的主要原因有:第一,煅烧时氯铂酸分解产生HCl和Cl2,这使复合催化剂的比表面积大幅度增加,从而增加催化剂对污染物的吸附性能和提供更多的活性位点;第二,掺杂的Pt2+/Pt0可充当电子库,捕捉从激发的g-C3N4中迁移的电子,抑制了g-C3N4产生的电子和空穴的复合;第三,掺杂适当量的Pt2+/Pt0可以提升g-C3N4对光的吸收,从而产生更多的自由基参与光催化的降解反应。
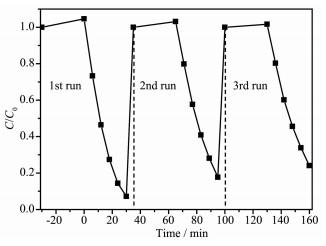
为了考察Pt2+/Pt0-CN的光催化稳定性,我们选取Pt0.02-CN进行循环使用的稳定性测试。结果如图 9所示。每次运行RhB的降解率仅有少量下降,说明Pt0.02-CN具有比较稳定的光催化活性。
为了测定光催化反应的活性物种,利用对苯醌( p-BZQ)、乙二胺四乙酸二钠(EDTA-2Na)、叔丁醇(TBA)分别作为·O2-自由基、空穴(h+)和OH自由基的捕获剂,进行光催化反应的自由基捕获效应研究。在降解10 mg·L-1 RhB时,添加捕获剂的量为1 mmol·L-1,其它实验条件与不加捕获剂的光催化活性测试条件保持相同。测试结果表明,加入对苯醌、乙二胺四乙酸二钠、叔丁醇后,Pt0.02-CN对RhB的降解率从84%下降到19%、80%,和78%,由此说明·O2-自由基是降解过程中的主要活性物种。
以三聚氰胺和氯铂酸溶液为原料,采用共缩聚法制备了Pt含量很低的Pt2+/Pt0-g-C3N4复合光催化剂。发现掺杂微量Pt可较大幅度增大g-C3N4的比表面积,同时提升光吸收性能和光生电子和空穴的分离能力。·O2-自由基是降解RhB过程中的主要活性物种。制备获得的Pt2+/Pt0-g-C3N4复合光催化剂在降解偶氮染料和抗生素具有较高的活性。Pt0.02-CN对环丙沙星、罗丹明B、酸性橙Ⅱ、甲基橙和亚甲基蓝的降解效率分别比g-C3N4提升了43%、64%、39%、42%和52%。本工作对探索制备低含量的贵金属掺杂光催化剂具有一定的借鉴作用。
Moon J, Yun C Y, Chung K W, et al. Catal. Today, 2003, 87(1):77-86
刘仁月, 吴榛, 白羽, 等.无机化学学报, 2017, 33(3):519-527 doi: 10.11862/CJIC.2017.062LIU Ren-Yue, WU Zhen, BAI Yu, et al. Chinese J. Inorg. Chem., 2017, 33(3):519-527 doi: 10.11862/CJIC.2017.062
Zeng D B, Yang K, Yu C L, et al. Appl. Catal. B-Environ., 2018, 227:449-463
Xing W N, Tu W G, Ou M, et al. ChemSusChem, 2019, 12(9):2029-2034 doi: 10.1002/cssc.201801431
Lu Z, Song W L, Ouyang C, et al. RSC Advances, 2017, 7(53):33552-33557 doi: 10.1039/C7RA04931E
Nagajyothi P C, Pandurangan M, Vattikuti S V P, et al. Sep. Purif. Technol., 2017, 188:228-237 doi: 10.1016/j.seppur.2017.07.026
Zhang W, Zhou L, Deng H P, et al. J. of Mol. Catal. A-Chem., 2016, 423:270-276 doi: 10.1016/j.molcata.2016.07.021
Sun T, Jiang H Y, Ma C C, et al. Catal. Commun., 2016, 79:45-48 doi: 10.1016/j.catcom.2016.03.004
Li K X, Zeng Z X, Yan L S, et al. Appl. Catal. B-Environ., 2015, 165:428-437 doi: 10.1016/j.apcatb.2014.10.039
Zhang P Y, Wang T T, Zeng H P. Appl. Surf. Sci., 2017, 391:404-414 doi: 10.1016/j.apsusc.2016.05.162
Cao S W, Jiang J, Zhu B C, et al. Phys. Chem. Chem. Phys., 2016, 18(28):19457-19463 doi: 10.1039/C6CP02832B
Wang X C, Kazuhiko M, Arne T, et al. Nature Mater., 2009, 8(1):76-80 doi: 10.1038/nmat2317
Ou M, Wan S P, Zhong Q, et al. Hydrogen Energy., 2017, 42(44):27043-27054 doi: 10.1016/j.ijhydene.2017.09.047
Li S L, Yin H, Kan X, et al. Phys. Chem. Chem. Phys., 2017, 19(44):30069-30077 doi: 10.1039/C7CP05195F
Hong Y P, Zhang J, Huang F, et al. J. Mater. Chem., 2015, 3(26):13913-13919 doi: 10.1039/C5TA02500A
Yu H G, Huang Y, Gao D D, et al. Ceram. Int., 2019, 45(8):9807-9813 doi: 10.1016/j.ceramint.2019.02.018
Jin X X, Zhang L X, Fan X Q, et al. Appl. Catal. B-Environ., 2018, 237:888-894 doi: 10.1016/j.apcatb.2018.06.035
Zhu Y Q, Wang T, Xu T, et al. Appl. Surf. Sci., 2019, 464:36-42 doi: 10.1016/j.apsusc.2018.09.061
Chen Y L, Li J H, Hong Z H, et al. Phys. Chem. Chem. Phys., 2014, 16(17):8106-8113 doi: 10.1039/c3cp55191a
Wang X J, Wang Q, Li F T, et al. Chem. Eng. J., 2013, 234:361-371 doi: 10.1016/j.cej.2013.08.112
Zhang D F, Meng X, Meng Y, et al. Sep. Purif. Technol., 2017, 186:282-289 doi: 10.1016/j.seppur.2017.06.026
Xiao J D, Xie Y B, Faheem N, et al. Appl. Catal. B-Environ., 2016, 183:417-425 doi: 10.1016/j.apcatb.2015.11.010
Han Q, Wang B, Gao J, et al. ACS NANO, 2016, 10(2):2745-2751 doi: 10.1021/acsnano.5b07831
Dong G H, Ai Z H, Zhang L Z. RSC Advances, 2015, 4(11):5553-5560
Li J H, Shen B, Hong Z H, et al. Chemi. Commun., 2012, 48(98):12017-12019 doi: 10.1039/c2cc35862j
Gao G P, Jiao Y, Eric R W, et al. J. Am. Chem. Soc., 2016, 138(19):6292-6297 doi: 10.1021/jacs.6b02692
Zhang S Q, Yang Y X, Guo Y N, et al. J. Hazard. Mater., 2013, 261:235-245 doi: 10.1016/j.jhazmat.2013.07.025
Chai B, Peng T Y, Mao J, et al. Phys. Chem. Chem. Phys., 2012, 14(48):16745-16752 doi: 10.1039/c2cp42484c
Wu J H, Shao F Q, Han S Y, et al. J. Colloid Interf. Sci., 2019, 535:41-49 doi: 10.1016/j.jcis.2018.09.080
Zhu Y Z, Xu Z X, Lang Q Q, et al. Appl. Catal. B-Environ., 2017, 206:282-292 doi: 10.1016/j.apcatb.2017.01.035
Zeng Z X, Li K X, Wei K, et al. Chin. J. Catal., 2017, 38(1):29-38 doi: 10.1016/S1872-2067(16)62589-5
陈范云, 张萌迪, 马小帅, 等.无机化学学报, 2019, 35(6):1034-1040 doi: 10.11862/CJIC.2019.115CHEN Fan-Yun, ZHANG Meng-Di, MA Xiao-Shuai, et al. Chinese J. Inorg. Chem., 2019, 35(6):1034-1040 doi: 10.11862/CJIC.2019.115
Ou M, Zhong Q, Zhang S L, et al. J. Alloys Compd., 2015, 626:401-409 doi: 10.1016/j.jallcom.2014.11.148
Wang W G, Yu J G, Xiang Q J, et al. Appl. Catal. B-Environ., 2012, 119:109-116
Liang S H, Zhang D F, Pu X P, et al. Sep. Purif. Technol., 2019, 210:786-797 doi: 10.1016/j.seppur.2018.09.008

图 2 g-C3N4和Pt2+/Pt0-CN样品的吸附-脱附等温线和孔径分布曲线
Figure 2 Adsorption-desorption isotherm and aperture distribution curve of the g-C3N4 and Pt2+/Pt0-CN samples

图 3 不同样品的扫描电镜照片: (a) g-C3N4; (b) Pt0.02-CN; 透射电镜照片: (c), (d) Pt0.02-CN; (e) g-C3N4
Figure 3 SEM images: (a) g-C3N4; (b) Pt0.02-CN; TEM images: (c), (d) Pt0.02-CN; (e) g-C3N4

图 4 (a) g-C3N4和Pt2+/Pt0-CN的紫外-可见漫反射光谱图; (b) g-C3N4和Pt2+/Pt0-CN的带隙值图
Figure 4 (a) UV-visible diffuse reflectance spectra of g-C3N4 and Pt2+/Pt0-CN the samples; (b) Band gap value graph of g-C3N4 and Pt2+/Pt0-CN samples

图 5 g-C3N4和Pt2+/Pt0-CN样品的红外谱图
Figure 5 Infrared spectra of the g-C3N4 and Pt2+/Pt0-CN samples

图 6 Pt0.02-CN样品的XPS谱: (a)全谱图; (b) C1s; (c) N1s; (d) Pt4f; (e) O1s
Figure 6 XPS spectra of the Pt0.02-CN sample: (a) Full spectrum; (b) C1s; (c) N1s; (d) Pt4f; (e) O1s

图 7 g-C3N4和Pt2+/Pt0-CN样品的荧光测试(a),光电流测试(b)和电化学阻抗测试(c)
Figure 7 Fluorescence test of g-C3N4 and Pt2+/Pt0-CN samples (a), Photocurrent test (b) and Electrochemical impedance test (c)

图 8 g-C3N4和Pt2+/Pt0-CN样品的光催化性能测试: (a) 10 mg·L-1甲基橙; (b) 10 mg·L-1罗丹明B; (c) 10 mg·L-1酸性橙Ⅱ; (d) 15 mg·L-1亚甲基蓝; (e) 10 mg·L-1环丙沙星
Figure 8 Photocatalytic performance test of g-C3N4 and Pt2+/Pt0-CN samples: (a) 10 mg·L-1 methyl orange; (b) 10 mg·L-1 RhB; (c) 10 mg·L-1 acidic orangeⅡ; (d) 15 mg·L-1 methylene blue; (e) 10 mg·L-1 ciprofloxacin

图 9 Pt0.02-CN降解10 mg·L-1 RhB的循环实验
Figure 9 Recycling test of Pt0.02-CN for the degradation of 10 mg·L-1 RhB
表 1 g-C3N4和Pt2+/Pt0-CN样品的比表面积、孔体积和孔尺寸
Table 1. Specific surface area, pore volume and pore size of the g-C3N4 and Pt2+/Pt0-CN samples
| Sample | Specific surface area / (m2 · g-1) | Pore volume / (cm3·g-1) | Pore size / nm |
| g-C3N4 | 4 | 0.04 | 34 |
| Pt0.002-CN | 27 | 0.12 | 18 |
| Pt0.02-CN | 32 | 0.10 | 12 |
| Pt0.1-CN | 20 | 0.07 | 14 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
