引用本文:
赵娣, 刘洪燕, 李桂花, 刘宇阳, 崔子硕. 可见光高催化活性GO/Ag3PO4/Ni复合薄膜的制备和性能[J]. 无机化学学报,
2020, 36(2): 253-260.
doi:
10.11862/CJIC.2020.012 Citation:
ZHAO Di, LIU Hong-Yan, LI Gui-Hua, LIU Yu-Yang, CUI Zi-Shuo. Preparation and Properties of Graphene-Oxide/Ag3PO4/Ni Composite Films with High Photocatalytic Activity under Visible Light[J]. Chinese Journal of Inorganic Chemistry,
2020, 36(2): 253-260.
doi:
10.11862/CJIC.2020.012
Received Date:
25 July 2019 Revised Date:
22 October 2019 Available Online:
01 February 2020
Abstract:
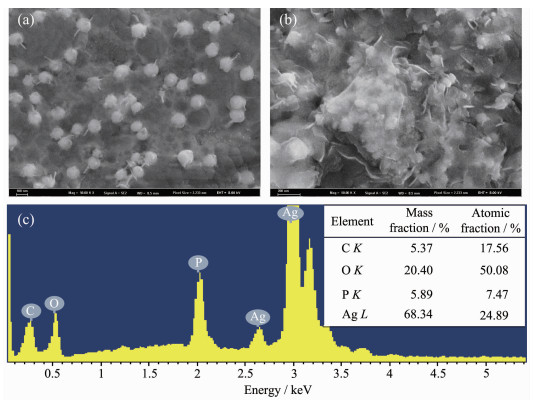
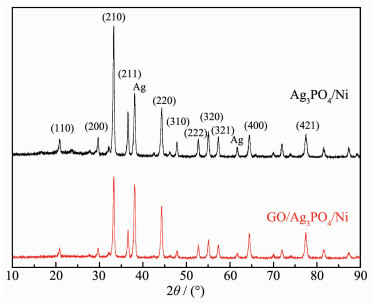
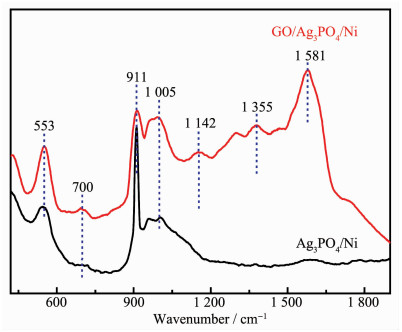
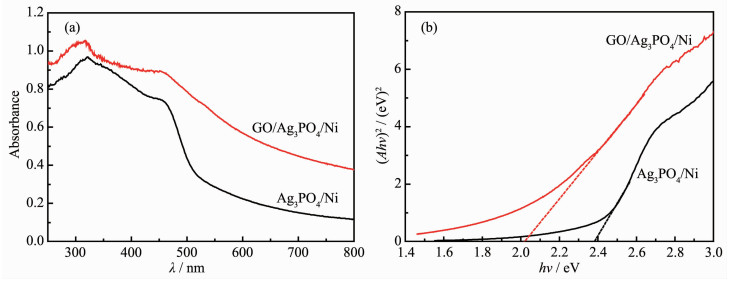
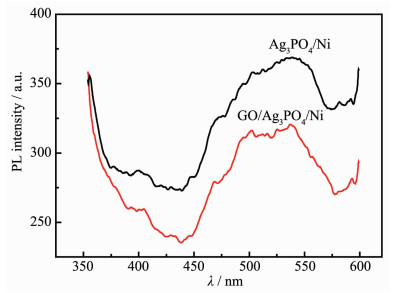
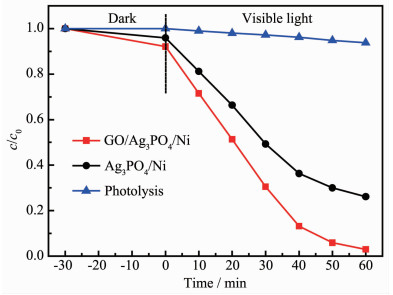
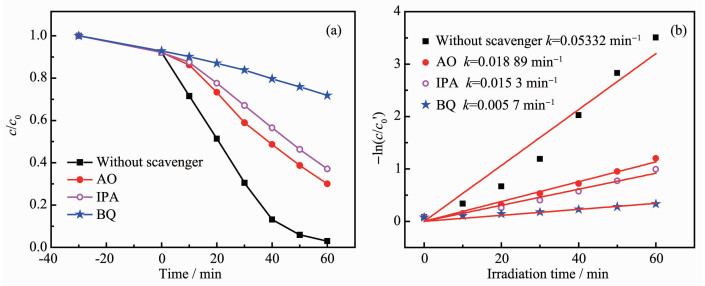
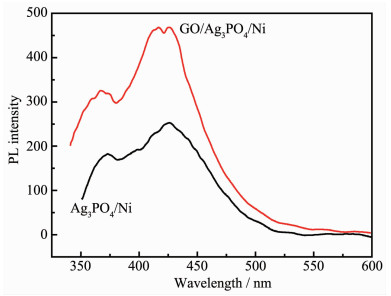
Ag3PO4 based-GO/Ag3PO4/Ni composite thin films were prepared by electrochemical co-deposition using a mixture of ammonium phosphate and graphene oxide aqueous suspension as electrolyte. Scanning electron microscopy (SEM), energy dispersive spectroscopy (EDS), X-ray diffraction (XRD), Raman and ultraviolet-visible diffuse reflectance spectroscopy (UV-Vis DRS) were employed to analyze the morphology, crystallinity and optical characteristics of the composite films. The results showed that the GO/Ag3PO4/Ni composite films prepared by the optimum process exhibited GO-coated surface morphology outside Ag3PO4 nanospheres with a diameter of about 100 nm. The strong charge interactions existed between GO sheets and Ag3PO4 nanospheres. As compared to Ag3PO4 nanospheres alone, the attachments of GO sheets led to a band gap narrowing and a strong absorbance in visible region. The photocatalytic (PC) activity and stability of the GO/Ag3PO4/Ni thin films were investigated by following the degradation of rhodamine B (RhB) under visible light irradiation. The PC mechanism of the composite films was explored by fluorescence spectroscopy and trapping agent method. Notably, the incorporation of GO sheets not only significantly enhanced the PC activity but also improved the structural stability of Ag3PO4. The efficiency of PC degradation of RhB under visible light irradiation 60 min was 1.32 times greater than that observed when a Ag3PO4/Ni thin film was used. GO/Ag3PO4/Ni thin films could be reused seven times without any significant decrease in the catalytic activity of the film. The excellent charge conductivity of GO and the positive synergistic effect between Ag3PO4 nanospheres and GO sheets are proposed to contribute to the improved PC properties of the composite films.
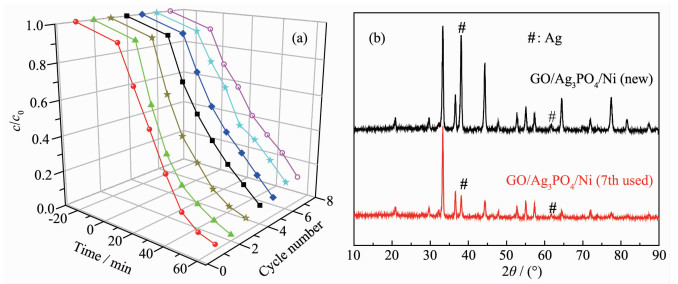
(a) Repeated photocatalytic degradation of RhB solution under visible light irradiation; (b) XRD patterns of the fresh GO/Ag3PO4/Ni thin films and the used GO/Ag3PO4/Ni thin films after seven recycling runs
李爱昌, 李桂花, 冀晓燕, 等.无机化学学报, 2017, 33(12):2247-2254 doi: 10.11862/CJIC.2017.277LI Ai-Chang, LI Gui-Hua, JI Xiao-Yan, et al. Chinese J. Inorg. Chem., 2017, 33(12):2247-2254 doi: 10.11862/CJIC.2017.277
[11]
Zhang L L, Zhang H C, Huang H, et al. New J. Chem., 2012, 36(8):1541-1544 doi: 10.1039/c2nj40206h
[12]
Yao W F, Zhang B, Huang C P, et al. J. Mater. Chem., 2012, 22:4050-4055 doi: 10.1039/c2jm14410g
(a) Repeated photocatalytic degradation of RhB solution under visible light irradiation; (b) XRD patterns of the fresh GO/Ag3PO4/Ni thin films and the used GO/Ag3PO4/Ni thin films after seven recycling runs

 下载:
下载:

 下载:
下载:










