Table 1.
Crystal data and structure refinements for complexes 1~2
Citation:
LIU Zhi-Qiang, WU Jun-Feng, CHEN Jun, WU Xia, WANG Yan. Two Metal-Organic Frameworks Constructed by 1, 3-Di(1H-imidazol-4-yl) Ligand: Synthesis, Crystal Structure and Photoluminescence Property[J]. Chinese Journal of Inorganic Chemistry,
2020, 36(1): 159-164.
doi:
10.11862/CJIC.2020.003

1, 3-二(4-咪唑基)苯构筑的两个金属-有机框架化合物:合成、晶体结构和荧光性质
摘要:
利用1,3-二(4-咪唑基)苯(L)和过渡金属盐反应,通过水热法方法合成了2个新颖的金属有机框架化合物:[Cd(L)(SO4)]n(1)和{[Ni(L)(SO4)]·H2O}n(2)。利用元素分析、红外、粉末衍射、热重分析等对MOFs进行了表征。通过X射线单晶衍射表明:1属于正交晶系,Pnma空间群,a=0.707 28(5)nm,b=1.343 81(9)nm,c=1.407 31(9)nm,V=1.337 58(16)nm3,Dc=2.079 g·cm-3,Z=4,F(000)=824,GOF=1.072,R1=0.019 0,wR2=0.050 6。2属于三斜晶系,P1空间群,a=0.986 65(18)nm,b=1.145 7(2)nm,c=1.302 5(2)nm,α=65.029(3)°,β=83.497(3)°,γ=86.423(4)°,V=1.326 0(4)nm3,Dc=1.441 g·cm-3,Z=2,F(000)=592,GOF=1.016,R1=0.043 4,wR2=0.077 1。2个MOFs均为二维的层状结构。此外,对MOFs的热稳定性和荧光性质也进行了研究。
-
关键词:
- 金属-有机框架化合物
- / 双4-咪唑配体
- / 晶体结构
- / 荧光性质
English
Two Metal-Organic Frameworks Constructed by 1, 3-Di(1H-imidazol-4-yl) Ligand: Synthesis, Crystal Structure and Photoluminescence Property
Abstract:
Reactions of ligands 1, 3-di(1H-imidazol-4-yl)benzene (L) and with corresponding metal salts under hydrothermal conditions gave rise to two new metal-organic frameworks (MOFs), namely[Cd(L)(SO4)]n (1) and {[Ni(L)(SO4)]·H2O}n (2). All of MOFs have been structurally characterized by single-crystal X-ray diffraction analyses and characterized by elemental analysis, infrared spectra (IR), powder X-ray diffraction (PXRD) and thermo-gravimetric analysis (TGA). 1 belongs to orthorhombic system, space group Pnma, with a=0.707 28(5) nm, b=1.343 81(9) nm, c=1.407 31(9) nm, V=1.337 58(16) nm3, Dc=2.079 g·cm-3, Z=4, F(000)=824, GOF=1.072, R1=0.019 0, wR2=0.050 6. 2 belongs to triclinic system, space group P1, with a=0.986 65(18) nm, b=1.145 7(2) nm, c=1.302 5(2) nm, α=65.029(3)°, β=83.497(3)°, γ=86.423(4)°, V=1.326 0(4) nm3, Dc=1.441 g·cm-3, Z=2, F(000)=592, GOF=1.016, R1=0.043 4, wR2=0.077 1. All the MOFs have two-dimensional (2D) layer structures. Furthermore, the thermal stability and photoluminescence property of the MOFs were investigated.
-
0. Introduction
Benefiting from large surface areas, adjustable pore size, readily modifiable functional groups and enough active sites, metal-organic frameworks (MOFs) are widely used in the fields of gas adsorption and separation, drug delivery, sensing and detection, catalysis and so on[1-7]. It is well-known that the potential applications of MOFs largely depend on their structural characteristics. Due to MOFs syntheses affected by many factors including organic ligands, solvent systems, pH, metal ions and so on, so it still has a large challenge the syntheses of target MOFs. Of all these conditions, the organic ligand is of vital aspect because physical properties of ligands directly affect the frameworks and properties of MOFs[8-12]. Our previous studies have demonstrated that organic compounds with 1H-imidazol-4-yl groups are versatile ligands for the construction of MOFs with definite structures and properties[13-16].
Herein we focused our attention on reactions of 1, 3-di(1H-imidazol-4-yl)benzene (L) with Cd(Ⅱ) or Ni(Ⅱ) salts. In this work, two MOFs were successfully synthesized, namely [Cd(L)(SO4)]n (1) and {[Ni(L)(SO4)]·H2O}n (2). The MOFs were characterized by X-ray crystallography, IR spectroscopy, elemental and thermal analyses. In addition, photoluminescence property of the MOF 1 was investigated.
1. Experimental
1.1 Materials
and methods All commercially available chemicals and solvents are of reagent grade and were used as received without further purification. The ligand L was synthesized according to the procedure reported in the literature[17]. FT-IR spectra were recorded in a range of 400~4 000 cm-1 on a Bruker Vector 22 FT-IR spectro-meter using KBr pellets. Thermogravimetric analysis (TGA) was performed on a Mettler-Toledo (TGA/DSC1) thermal analyzer under nitrogen with a heating rate of 10 ℃·min-1. Powder X-ray diffraction (PXRD) patterns were obtained on a Bruker D8 Advance diffractometer using Cu Kα (λ=0.154 18 nm) in 2θ range of 5°~50° at 293 K, in which the X-ray tube was operated at 40 kV and 40 mA. Photoluminescence spectra were measured on an Aminco Bowman Series 2 spectrofluorometer with a xenon arc lamp as the light source. In the measurements of emission and excitation spectra, the pass width is 5 nm.
1.2 Synthesis of complexes 1 and 2
1.2.1 Synthesis of complex 1
3CdSO4·8H2O (17.6 mg, 0.05 mmol) and L (10.5 mg, 0.05 mmol) were dissolved in H2O (10 mL) was sealed in a Teflon-lined stainless steel container and heated at 90 ℃ for 3 days. The resulting colorless block crystals were collected in 68% yield based on L. Elemental analysis Calcd. for C12H10N4O4SCd(%): C 34.42, H 2.41, N 13.38; Found(%): C 34.47, H 2.53, N 13.29. Selected IR peaks (cm-1, Fig.S1): 3 423 (s), 3 108 (s), 2 880 (s), 1 620 (w), 1 586 (w), 1 497 (m), 1 366 (w), 1 126 (s), 853 (s), 794 (s), 603 (s).
1.2.2 Synthesis of complex 2
The synthesis was similar with that of 1 except that NiSO4·6H2O (13.1 mg, 0.05 mmol) was used instead of 3CdSO4·8H2O. The resulting green block crystals were collected in 82% yield based on L. Elemental analysis Calcd. for C24H22N8O5SNi(%): C 48.59, H 3.74, N 18.89; Found(%): C 48.53, H 3.83, N 18.82. Selected IR peaks (cm-1, Fig.S1): 3 427 (s), 2 855 (s), 1 623 (m), 1 581 (m), 1 449 (m), 1 245 (s), 1 120 (s), 1 092 (s), 983 (s), 830 (m), 747 (w).
1.3 Crystallographic data collection and refinement
Crystallographic data for 1 and 2 were collected on a Bruker Smart Apex Ⅱ CCD with graphite monochromated Mo Kα radiation source (λ=0.710 73 nm). The diffraction data as well as the intensity corrections for the Lorentz and polarization effects, was carried out using the SAINT program[18]. Semi-empirical absorption correction was applied using SADABS program[19]. The structures of 1 and 2 were determined by direct methods and refined anisotro-pically on F 2 by the full-matrix least-squares technique with SHELXTL-2016 program package[20-21]. The hydro-gen atoms except for those of water molecules were generated geometrically and refined isotropically using the riding model. For 2, the SQUEEZE subroutine of the PLATON software suite was used to remove the scattering from the highly disordered solvent molecules[22]. The final formulas were calculated from the SQUEEZE results, TGA and elemental analysis. Selected bond lengths and angles are listed in Table S1 (Supporting information). The hydrogen bonding data are provided in Table S2.
CCDC: 1920722, 1; 1920723, 2.
Table 1

 下载:
导出CSV
下载:
导出CSV
Complex 1 2 Empirical formula C12H10N4O4SCd C24H22N8O5SNi Formula weight 418.70 593.24 Crystal system Orthorhombic Triclinic Space group Pnma P1 a / nm 0.707 28(5) 0.986 65(18) b / nm 1.343 81(9) 1.145 7(2) c / nm 1.407 31(9) 1.302 5(2) α / (°) 65.029(3) β / (°) 83.497(3) γ / (°) 86.423(4) V / nm3 1.337 58(16) 1.326 0(4) Z 4 2 Dc / (g·cm-3) 2.079 1.441 μ / mm-1 1.814 0.856 F(000) 824 592 Reflection collected 4 980 9 345 Unique reflection 1 235 4 641 Goodness-of-fit 1.072 1.016 R1a [I > 2σ(I)] 0.019 0 0.043 4 wR2b [I > 2σ(I)] 0.050 6 0.077 1 a R1=∑||Fo|-|Fc||/∑|Fo|; b wR2=|∑w(|Fo|2-|Fc|2)|/∑|w(Fo)2|1/2, where w=1/[σ2Fo2+(aP)2+bP], P=(Fo2+2Fc2)/3. 2. Result and discussion
2.1 Crystal structures of 1 and 2
2.1.1 Crystal structure of 1
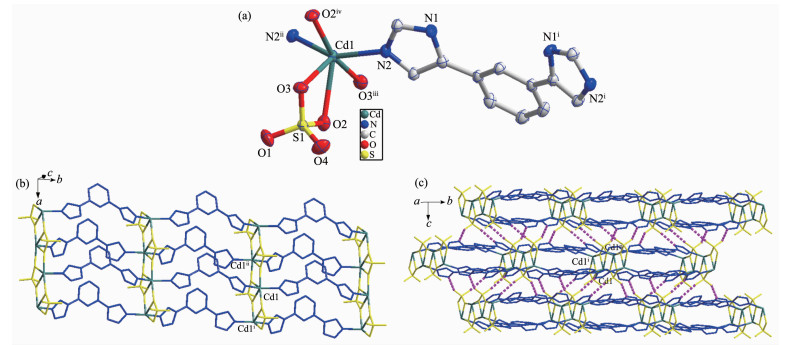
Single crystal X-ray structural analysis reveals that complex 1 crystallizes in the triclinic Pnma space group. As exhibited in Fig. 1a, the Cd(Ⅱ) is six-coordinated by four oxygen atoms (O1, O2, O3, O4) from the bridging sulfate anion and two N atoms (N2, N2ⅱ) from distinct L. The asymmetric unit of 1 contains one molecule of [Cd(L)(SO4)]. The Cd-O bond distances are from 0.236 2(2) to 0.253 7(3) nm, while the Cd-N ones are 0.218 39(19) and 0.218 40(19) nm. The coordination angles around Cd(Ⅱ) in 1 are from 57.09(7)° to 163.59(9)° (Table S1). Each L links two Cd(Ⅱ) to form an infinite one-dimensional (1D) chain, and the Cd-L 1D chains are further connected by sulfate anion to generate a 2D network (Fig. 1b), which is further extended into a three-dimensional (3D) supramolecular architecture through O-H…O hydrogen bonding interactions (Fig. 1c and Table S2).
Figure 1
 Figure 1. (a) Coordination environment of Ni(Ⅱ) ion in 2 with the ellipsoids drawn at 30% probability level; (b) 2D structure of 2; (c) 3D structure of 2 with hydrogen bonds indicated by dashed lines Fig. 3 TGA curves of 1 and 2
Figure 1. (a) Coordination environment of Ni(Ⅱ) ion in 2 with the ellipsoids drawn at 30% probability level; (b) 2D structure of 2; (c) 3D structure of 2 with hydrogen bonds indicated by dashed lines Fig. 3 TGA curves of 1 and 2Hydrogen atoms are omitted for clarity; Symmetry codes: ⅰx, y+1, z; ⅱx, y-1, z+1; ⅲ x, y+1, z-1; ⅳ x, y-1, z
2.1.2 Crystal structure of 2
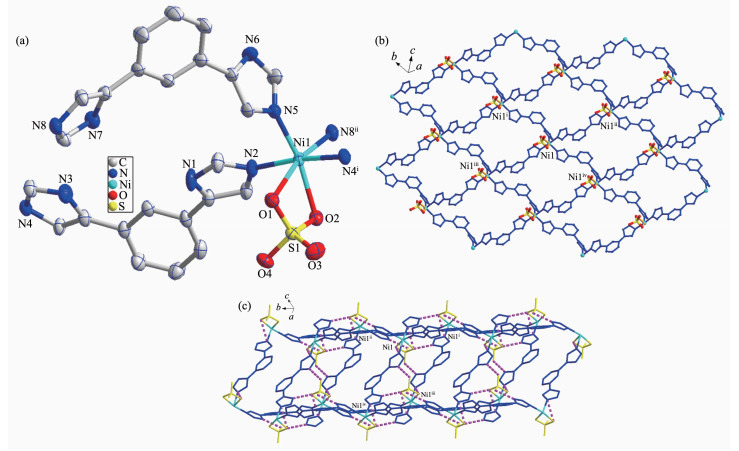
When NiSO4·6H2O instead of 3CdSO4·8H2O was used under the same reaction conditions for preparation of 1, complex 2 was obtained. The results of single crystal X-ray structural analysis showed that 2 crystallizes in triclinic space group P1. As shown in Fig. 2a, Ni1 is six-coordinated by four N atoms (N2, N4ⅰ, N5, N8ⅱ) from four different L ligands, two O atoms (O1, O2) from sulfate anion. The Ni-N bond distances are from 0.204 0(2) to 0.209 6(2) nm, while the Ni-O ones are 0.218 56(18) and 0.219 42(18) nm. The coordination angles around Ni(Ⅱ) in 1 are from 65.25(6)° to 172.49(9)° (Table S1). Each L links two Ni(Ⅱ) to generate a 2D network (Fig. 1b), which is further extended into a three-dimensional (3D) supra-molecular architecture through O-H…O hydrogen bonding interactions (Fig. 1c and Table S2).
Figure 2
 Figure 2. (a) Coordination environment of Ni(Ⅱ) ion in 2 with the ellipsoids drawn at 30% probability level; (b) 2D structure of 2; (c) 3D structure of 2 with hydrogen bonds indicated by dashed lines
Figure 2. (a) Coordination environment of Ni(Ⅱ) ion in 2 with the ellipsoids drawn at 30% probability level; (b) 2D structure of 2; (c) 3D structure of 2 with hydrogen bonds indicated by dashed linesHydrogen atoms are omitted for clarity; Symmetry codes: ⅰx, y+1, z; ⅱx, y-1, z+1; ⅲ x, y+1, z-1; ⅳ x, y-1, z
2.2 Powder X-ray diffraction and thermogravimetric analyses
The purity for the as-synthesized samples was ensured by PXRD measurements and the results are provided in Fig.S2. Each as-synthesized sample gives consistent PXRD pattern with the corresponding simulated one, indicating the pure phase of 1 and 2.
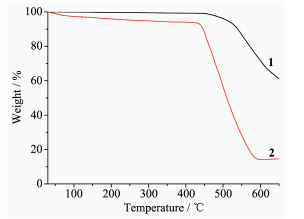
The thermal stability of 1 and 2 was examined by TGA in the N2 atmosphere from 30 to 650 ℃, and the TG curves are given in Fig. 3. The result was that MOF 1 did not show obvious weight loss before the decomposition of the framework at about 450 ℃, which is in agreement with the result of the crystal structure analysis. For MOF 2, the first weight loss of 3.1% from 30 to 130℃ is in accordance with the loss of free H2O molecules (Calcd. 3.03%), and the framework maintained before about 430 ℃.
Figure 3
2.3 Photoluminescence property
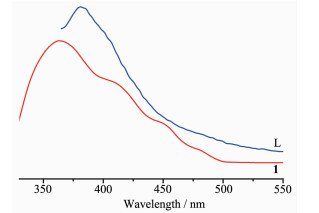
It is known that the frameworks with d10 metal centers and π-conjugated organic ligands may have photoluminescence. Luminescence in MOFs generally arises from the building components: conjugated organic ligands and/or metal ions or clusters. Organic linkers with aromatic moieties or extended π systems are commonly used in the construction of porous MOFs due to their rigid molecular backbone. MOFs with d10 metal centers show luminescent properties and is likely to be candidates for luminescent materials[23-29]. Accordingly, the solid-state luminescent emission spectra of L and 1 were collected at room temperature. As shown in Fig. 4, the free ligand L exhibited an emission maxima at 381 nm (λex=335 nm), and MOF 1 gave emission maxima at 363 nm (λex=330 nm). Compared with the emission of free L, the blue-shifts of the emissions of 1 are considered to be caused by the coordination of the ligand to the metal centers.
Figure 4
 Figure 4. Emission spectra of 1 as well as free L ligand in solid state at room temperature
Figure 4. Emission spectra of 1 as well as free L ligand in solid state at room temperature3. Conclusions
In summary, two MOFs based on 1, 3-di(1H-imidazol-4-yl)benzene (L) and corresponding metal salts were synthesized and characterized. The structures of them are 2D network, which are further linked together by hydrogen bonds to give eventual 3D architectures. Furthermore, the thermal stability and photolumine-scence property of the MOFs were investigated.
Supporting information is available at http://www.wjhxxb.cn
-
-
[1]
Kang Y S, Lu Y, Chen K, et al. Coord. Chem. Rev., 2019, 378:262-280 doi: 10.1016/j.ccr.2018.02.009
-
[2]
Wu S Y, Min H, Shi W, et al. Adv. Mater., 2019, 31:180581-180584
-
[3]
Seoane B, Coronas J, Gascon I, et al. Chem. Soc. Rev., 2015, 44:2421-2454 doi: 10.1039/C4CS00437J
-
[4]
Zhang J P, Zhang Y B, Lin J B, et al. Chem. Rev., 2012, 112:1001-1033 doi: 10.1021/cr200139g
-
[5]
He C B, Liu D M, Lin W B. Chem. Rev., 2015, 115:11079-11108 doi: 10.1021/acs.chemrev.5b00125
-
[6]
Cook T R, Zheng Y R, Stang P J. Chem. Rev., 2013, 113:734-777 doi: 10.1021/cr3002824
-
[7]
Tanabe K K, Cohen S M. Chem. Soc. Rev., 2011, 40:498-519 doi: 10.1039/C0CS00031K
-
[8]
Lan Y Q, Jiang H L, Li S L, et al. Inorg. Chem., 2012, 51:7484-7491 doi: 10.1021/ic202635a
-
[9]
Lin Z J, Lu J, Hong M C, et al. Chem. Soc. Rev., 2014, 43:5867-5895 doi: 10.1039/C3CS60483G
-
[10]
Sun Y X, Sun W Y. CrysEngComm, 2015, 17:4045-4063 doi: 10.1039/C5CE00372E
-
[11]
Wang H, Yi F Y, Dang S, et al. Cryst. Growth Des., 2014, 14:147-156 doi: 10.1021/cg4013334
-
[12]
Wu H, Liu H Y, Yang J, et al. Chem. Commun., 2011, 47:1818-1820 doi: 10.1039/C0CC04724D
-
[13]
Liu Z Q, Zhao Y, Wang P, et al. Dalton. Trans., 2017, 46:9022-9029 doi: 10.1039/C7DT01759F
-
[14]
Liu Z Q, Zhao Y, Zhang X D, et al. Dalton. Trans., 2017, 46:13943-13951 doi: 10.1039/C7DT02981K
-
[15]
Liu Z Q, Zhao Y, Deng Y, et al. Sens. Actuators B:Chem., 2017, 250:179-188 doi: 10.1016/j.snb.2017.04.151
-
[16]
Liu Z Q, Chen K, Zhao Y, et al. Cryst. Growth Des., 2018, 18:1136-1146 doi: 10.1021/acs.cgd.7b01572
-
[17]
Have R T, Huisman M, Meetsma A, et al. Tetrahedron, 1997, 53:11355-11368 doi: 10.1016/S0040-4020(97)00717-5
-
[18]
SAINT, Program for Data Extraction and Reduction, Bruker AXS, Inc., Madison, WI, 2001.
-
[19]
Sheldrick G M. SADABS, University of Göttingen, Germany, 2003.
-
[20]
Sheldrick G M. Acta Crystallogr. Sect. A:Found. Crystallogr., 2015, A71:3-8
-
[21]
Sheldrick G M. Acta Crystallogr. Sect. C:Struct. Chem. Commun., 2015, C71:3-8
-
[22]
Spek A L. PLATON, A Multipurpose Crystallographic Tool, Utrecht University, The Netherlands, 2013.
-
[23]
Hu Z C, Deibert B J, Li J. Chem. Soc. Rev., 2014, 43:5815-5840 doi: 10.1039/C4CS00010B
-
[24]
Dai J C, Wu X T, Fu S M, et al. Chem. Commun., 2002:12-13
-
[25]
Chen W, Wang J Y, Chen Q, et al. Inorg. Chem., 2003, 42:944-946 doi: 10.1021/ic025871j
-
[26]
Ni J, Wei K J, Min Y, et al. Dalton Trans., 2012, 41:5280-5293 doi: 10.1039/c2dt12032a
-
[27]
Senchyk G A, Bukhan'ko V O, Lysenko A B, et al. Inorg. Chem., 2012, 51:8025-8033 doi: 10.1021/ic3000894
-
[28]
Liu Z Q, Zhao Y, Liu X H, et al. Polyhedron, 2019, 167:33-38 doi: 10.1016/j.poly.2019.04.007
-
[29]
刘志强, 黄永清, 孙为银.无机化学学报, 2017, 33(11):1959-1969 doi: 10.11862/CJIC.2017.244LIU Zhi-Qiang, HUANG Yong-Qing, SUN Wei-Yin. Chinese J. Inorg. Chem., 2017, 33(11):1959-1969 doi: 10.11862/CJIC.2017.244
-
[1]
-
Figure 1 (a) Coordination environment of Ni(Ⅱ) ion in 2 with the ellipsoids drawn at 30% probability level; (b) 2D structure of 2; (c) 3D structure of 2 with hydrogen bonds indicated by dashed lines Fig. 3 TGA curves of 1 and 2
Hydrogen atoms are omitted for clarity; Symmetry codes: ⅰx, y+1, z; ⅱx, y-1, z+1; ⅲ x, y+1, z-1; ⅳ x, y-1, z
Figure 2 (a) Coordination environment of Ni(Ⅱ) ion in 2 with the ellipsoids drawn at 30% probability level; (b) 2D structure of 2; (c) 3D structure of 2 with hydrogen bonds indicated by dashed lines
Hydrogen atoms are omitted for clarity; Symmetry codes: ⅰx, y+1, z; ⅱx, y-1, z+1; ⅲ x, y+1, z-1; ⅳ x, y-1, z
Figure 4 Emission spectra of 1 as well as free L ligand in solid state at room temperature
Table 1. Crystal data and structure refinements for complexes 1~2
Complex 1 2 Empirical formula C12H10N4O4SCd C24H22N8O5SNi Formula weight 418.70 593.24 Crystal system Orthorhombic Triclinic Space group Pnma P1 a / nm 0.707 28(5) 0.986 65(18) b / nm 1.343 81(9) 1.145 7(2) c / nm 1.407 31(9) 1.302 5(2) α / (°) 65.029(3) β / (°) 83.497(3) γ / (°) 86.423(4) V / nm3 1.337 58(16) 1.326 0(4) Z 4 2 Dc / (g·cm-3) 2.079 1.441 μ / mm-1 1.814 0.856 F(000) 824 592 Reflection collected 4 980 9 345 Unique reflection 1 235 4 641 Goodness-of-fit 1.072 1.016 R1a [I > 2σ(I)] 0.019 0 0.043 4 wR2b [I > 2σ(I)] 0.050 6 0.077 1 a R1=∑||Fo|-|Fc||/∑|Fo|; b wR2=|∑w(|Fo|2-|Fc|2)|/∑|w(Fo)2|1/2, where w=1/[σ2Fo2+(aP)2+bP], P=(Fo2+2Fc2)/3.  下载: 导出CSV
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 2
- 文章访问数: 466
- HTML全文浏览量: 34
