引用本文:
杨文琦, 汪杰, 乔园园, 王贵昌. 金属单原子模型催化剂热稳定性的反应力场(ReaxFF)分子动力学研究[J]. 无机化学学报,
2019, 35(11): 2078-2082.
doi:
10.11862/CJIC.2019.251 Citation:
YANG Wen-Qi, WANG Jie, QIAO Yuan-Yuan, WANG Gui-Chang. Thermal Stability of Single Atom Metal Catalysts: ReaxFF Molecular Dynamics Study[J]. Chinese Journal of Inorganic Chemistry,
2019, 35(11): 2078-2082.
doi:
10.11862/CJIC.2019.251
Key Laboratory of Advanced Energy Materials Chemistry(Ministry of Education) and Synergetic Innovation Center of Chemical Science and Engineering (Tianjin), College of Chemistry, Nankai University, Tianjin 300071, China
Received Date:
23 July 2019 Revised Date:
10 October 2019 Available Online:
10 November 2019
Abstract:
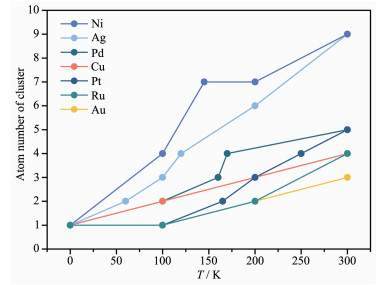
The catalytic activity of metal catalysts is closely related to its coordination unsaturation:The higher coordination unsaturation is, the better catalytic effect is in general. The single-atom catalyst (SAC or ad-atom) model has the smallest coordination number on metal surface and thus exhibits high catalytic activity, but the stability of SAC still need to be studied in detail. In this work, we used LAMMPS (large-scale atomic/molecular massively parallel simulator) for large-scale molecular dynamics simulation based on Reactive Force Field (ReaxFF) to study the thermal stability of the single atom model. The simulation results show that only Fe1/Fe(100) catalyst can be stable at high temperature (>500 K), while other SAC models will occur that single atom aggregates to form large nanoparticles or moves to subsurface as temperature increases. In the meanwhile, the dynamic behavior of Ni1/Ni(111) catalyst under H2 and O2 atmosphere was also studied was found that compared with simulation results in vacuum, the H2 and O2 atmosphere improved the stability of the catalyst to some extent.
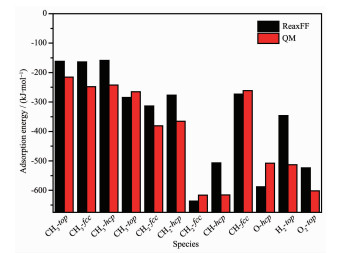
Figure 1.
ReaxFF fits for the binding energy of different adsorbed species (CH3, CH2, CH, O and H) on the different site of Ni(111) surface and H2, O2 molecular on the Ni1/Ni(111)
Figure 2.
Side view of dynamics configurations (100 ps) Cu, Ag, Au, Ni, Pd, Pt, Ru, Fe single metal model from top to bottom respectively and Fe1/Fe(100) model at 300, 400, 500 and 600 K
Figure 1
ReaxFF fits for the binding energy of different adsorbed species (CH3, CH2, CH, O and H) on the different site of Ni(111) surface and H2, O2 molecular on the Ni1/Ni(111)
Figure 2
Side view of dynamics configurations (100 ps) Cu, Ag, Au, Ni, Pd, Pt, Ru, Fe single metal model from top to bottom respectively and Fe1/Fe(100) model at 300, 400, 500 and 600 K

 下载:
下载:



 下载:
下载:






