State Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
Received Date:
03 September 2019 Revised Date:
09 October 2019 Available Online:
10 November 2019
Abstract:
Similar to single-molecular magnets (SMMs), single-molecule toroics (SMTs) are defined as molecules with toroidal magnetic bi-stability. This type of compounds is characterized by the "vortex" spatial distribution of the weakly coupled magnetic moments, resulting in zero total magnetic momentum, but a non-vanishing toroidal magnetic moment. SMTs offer broad application prospects in quantum computing and information storage, and can also be used as a multiferroic materials with magneto-electric coupling effect. Great effort has been paid in the synthesis of SMTs since the first observation of the SMT behavior of typical [Dy3] molecules, and researchers are committed to the investigation of SMTs with enhanced molecular toroidal magnetization. In this review, several representative examples with SMT behavior will be addressed to provide a down to date overview of the burgeoning SMT complexes reported in recent years. Accordingly, we aim to illuminate the factors governing the arrangement of toroidal magnetic moments and the synthetic strategy on designing SMTs, and ultimately enlighten the study of SMTs with enhanced toroidal magnetization.
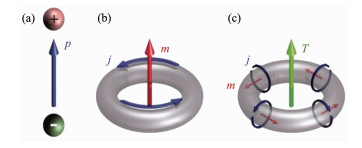
Figure 1.
(a) Electric (charge) dipole p created by pair of charges of opposite signs; (b) Magnetic dipole m produced by a current j flowing along a loop;
(c) Toroidal dipole T generated by currents flowing on a surface of a torus along its meridians (poloidal currents)[9]
Figure 3.
(a) Structure of [Dy3] with main anisotropy axes (dashed lines) and local magnetizations (arrows) in the ground state;
(b) Static magnetic properties of [Dy3] and their ab initio simulations (lines); (c) Two components of the ground Kramer′s doublet; (d) Dynamic magnetic susceptibility of [Dy3][31, 55]
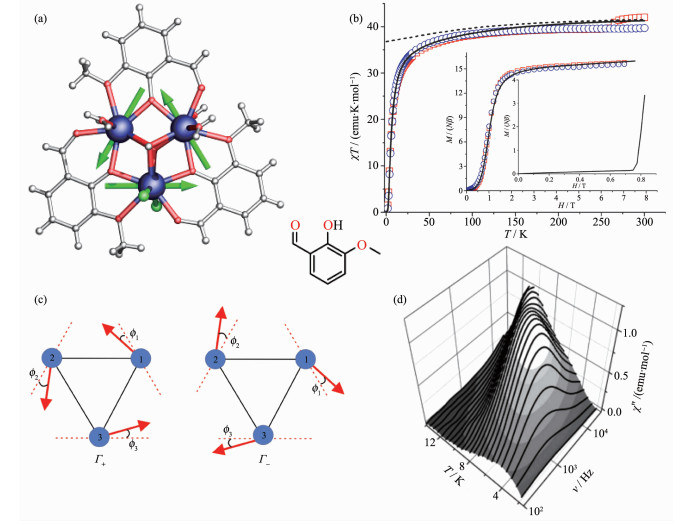
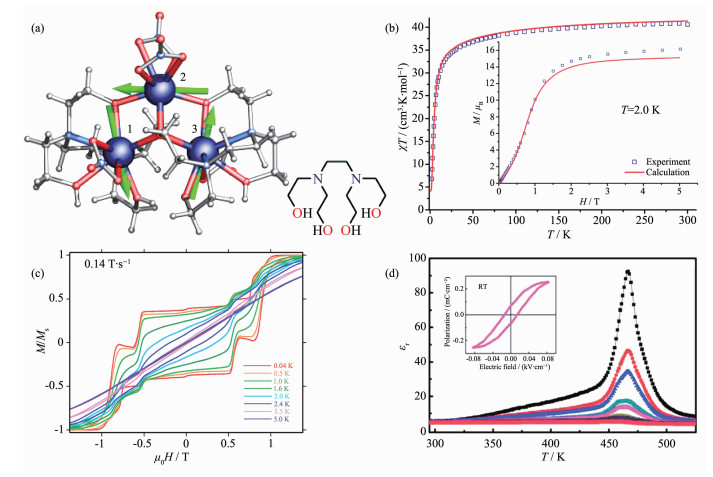
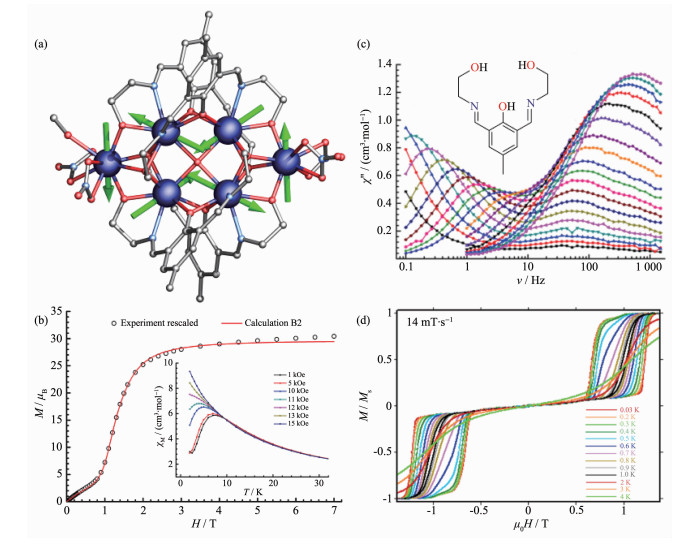
Figure 4.
(a) Structure with main anisotropy axes (dashed lines) and local magnetizations (arrows) in the ground state;
(b) Static magnetic properties of [Dy3]-1; (c) Hysteresis loops at indicated temperatures and sweep rate of 0.14 T·s-1 for [Dy3]-1; (d) Temperature dependence of the dielectric constant ε′(εr); Inset: dielectric hysteresis loops of [Dy3]-1[36]
Figure 5.
Crystal structure, static and dynamic magnetic properties of the first [Dy4] SMT, and the calculated anisotropic axes showing a toroidal alignment[37]
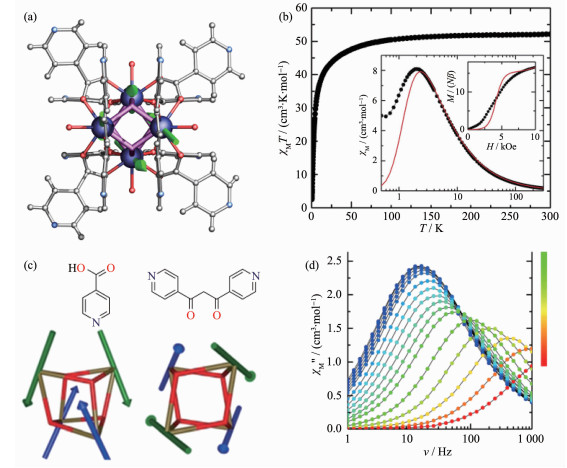
Figure 6.
(a) Representation of the crystal structure of [Dy4]-1; (b) Static magnetic properties of [Dy4]-1;
(c) Cubane core (dysprosium ions in blue and oxygen atoms in red) with the easy magnetic axes;
(d) Ac magnetic properties of [Dy4]-1[39]
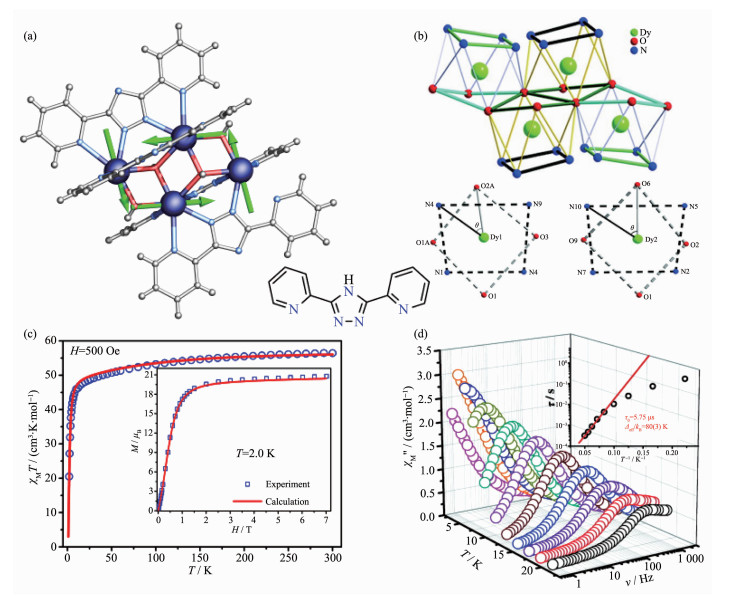
Figure 7.
(a) Structure of [Dy6]-1 with main anisotropy axes (dashed lines) and local magnetizations (arrows) in the ground state;
(b) Magnetic properties of [Dy6]-1[45]
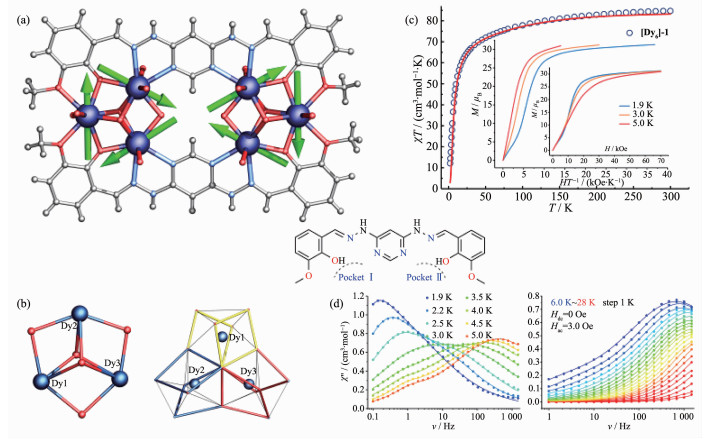
Figure 8.
(a, b) Structure of [Dy6]-2 with main anisotropy axes (dashed lines) and local magnetizations (arrows) in the ground state; (c, d) dc and ac magnetic properties of [Dy6]-2[46]
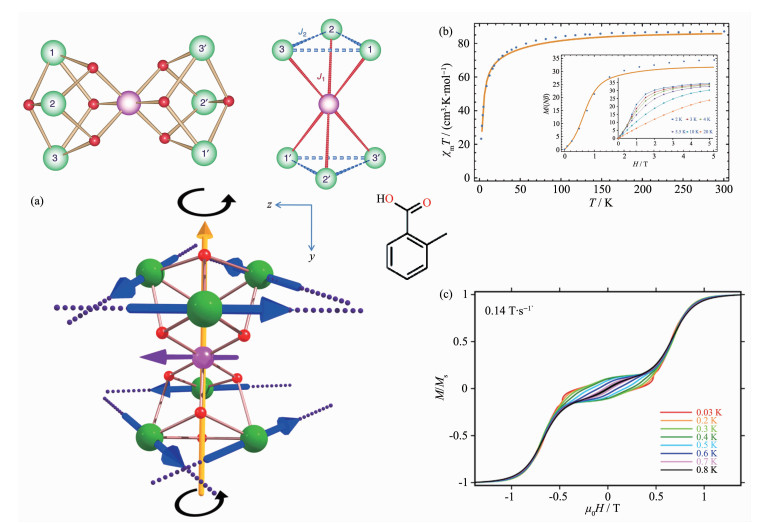
Figure 9.
(a) Structure of [CrDy6] (top), the directions of the local anisotropy axes in the ground Kramers doublet on each Dy site (dotted lines) in [CrDy6] (bottom); (b) χMT vs T plot and molar magnetization (M) vs magnetic field (H) at 1.9 K (Inset) for [CrDy6]; (c) Single-crystal magnetization (M) vs applied field measurements (μ-SQUID) for complex [CrDy6] at a range of 0.03~0.8 K with the scan rate of 0.14 T·s-1[48]
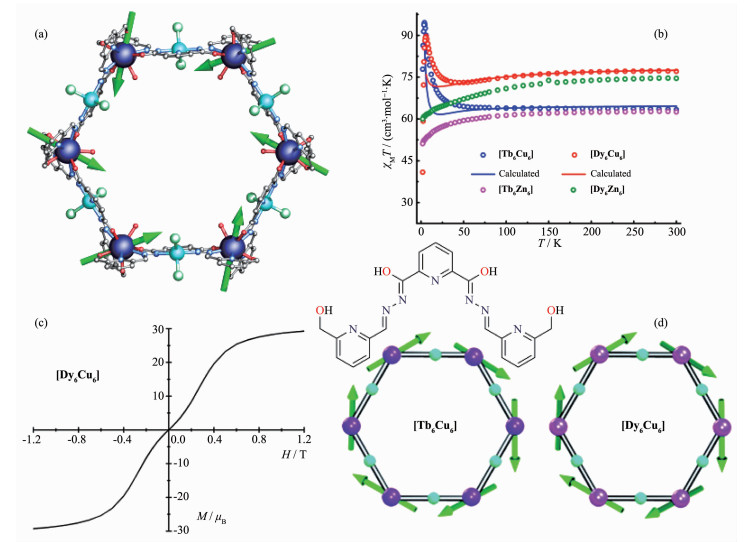
Figure 10.
(a) Structure of [Ln6Cu6] (Ln=Tb and Dy) with main anisotropy axes in the ground state; (b) χMT vs T plot for [Ln6Cu6] and [Ln6Zn6]; (c) Molar magnetization (M) vs magnetic field (H) at 1.9 K for [Dy6Cu6]; (d) Schematic drawings of 3d-4f[Ln6Cu6] (Ln=Tb and Dy) SMTs[51]
Figure 1
(a) Electric (charge) dipole p created by pair of charges of opposite signs; (b) Magnetic dipole m produced by a current j flowing along a loop;
(c) Toroidal dipole T generated by currents flowing on a surface of a torus along its meridians (poloidal currents)[9]
Figure 3
(a) Structure of [Dy3] with main anisotropy axes (dashed lines) and local magnetizations (arrows) in the ground state;
(b) Static magnetic properties of [Dy3] and their ab initio simulations (lines); (c) Two components of the ground Kramer′s doublet; (d) Dynamic magnetic susceptibility of [Dy3][31, 55]
Figure 4
(a) Structure with main anisotropy axes (dashed lines) and local magnetizations (arrows) in the ground state;
(b) Static magnetic properties of [Dy3]-1; (c) Hysteresis loops at indicated temperatures and sweep rate of 0.14 T·s-1 for [Dy3]-1; (d) Temperature dependence of the dielectric constant ε′(εr); Inset: dielectric hysteresis loops of [Dy3]-1[36]
Figure 5
Crystal structure, static and dynamic magnetic properties of the first [Dy4] SMT, and the calculated anisotropic axes showing a toroidal alignment[37]
Figure 6
(a) Representation of the crystal structure of [Dy4]-1; (b) Static magnetic properties of [Dy4]-1;
(c) Cubane core (dysprosium ions in blue and oxygen atoms in red) with the easy magnetic axes;
(d) Ac magnetic properties of [Dy4]-1[39]
Figure 7
(a) Structure of [Dy6]-1 with main anisotropy axes (dashed lines) and local magnetizations (arrows) in the ground state;
(b) Magnetic properties of [Dy6]-1[45]
Figure 8
(a, b) Structure of [Dy6]-2 with main anisotropy axes (dashed lines) and local magnetizations (arrows) in the ground state; (c, d) dc and ac magnetic properties of [Dy6]-2[46]
Figure 9
(a) Structure of [CrDy6] (top), the directions of the local anisotropy axes in the ground Kramers doublet on each Dy site (dotted lines) in [CrDy6] (bottom); (b) χMT vs T plot and molar magnetization (M) vs magnetic field (H) at 1.9 K (Inset) for [CrDy6]; (c) Single-crystal magnetization (M) vs applied field measurements (μ-SQUID) for complex [CrDy6] at a range of 0.03~0.8 K with the scan rate of 0.14 T·s-1[48]
Figure 10
(a) Structure of [Ln6Cu6] (Ln=Tb and Dy) with main anisotropy axes in the ground state; (b) χMT vs T plot for [Ln6Cu6] and [Ln6Zn6]; (c) Molar magnetization (M) vs magnetic field (H) at 1.9 K for [Dy6Cu6]; (d) Schematic drawings of 3d-4f[Ln6Cu6] (Ln=Tb and Dy) SMTs[51]

 下载:
下载:

 下载:
下载:











