Table 1.
Crystal data and structure refinement data of complexes 1~2
Citation:
HAN Xiao, SHAO Zhichao, ZHAO Bei, REN Ning, MENG Xiang-Ru, DING Jie, HOU Hong-Wei. Synthesis, Crystal Structure, Magnetic Properties and Catalytic Activity of Co(Ⅱ)/Ni(Ⅱ) Complexes with 3, 3'-(Pyridine-3, 5-diyl) Dibenzoic Acid[J]. Chinese Journal of Inorganic Chemistry,
2019, 35(11): 2159-2167.
doi:
10.11862/CJIC.2019.241

3, 3'-(吡啶-3, 5)二苯甲酸构筑的Co(Ⅱ)/Ni(Ⅱ)配合物的合成、晶体结构及性质
摘要:
设计、合成了2个配合物:{[Co2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n(1)和{[Ni2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n(2)(H2pddb=3,3'-(吡啶-3,5)二苯甲酸,DMA=N,N-二甲基乙酰胺),并通过红外光谱(IR),元素分析,热重分析(TG)和单晶X射线衍射确定它们的结构。结构分析表明1和2是异质同构的,其空间构型可简化成(3,6)连接的拓扑结构,其符号为(42·6)·(44·62·88·10)。进一步研究了配合物1和2的固体紫外、磁性及其催化性能。研究表明1和2中的中心金属离子之间存在反铁磁耦合作用,并且1可以作为C-H活化的高效非均相催化剂。
-
关键词:
- 3, 3'-(吡啶-3, 5)二苯甲酸
- / 配合物
- / 晶体结构
- / 磁性
- / 非均相催化
English
Synthesis, Crystal Structure, Magnetic Properties and Catalytic Activity of Co(Ⅱ)/Ni(Ⅱ) Complexes with 3, 3'-(Pyridine-3, 5-diyl) Dibenzoic Acid
Abstract:
Two complexes 1~2 (1={[Co2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n and 2={[Ni2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n, H2pddb=3, 3'-(pyridine-3, 5-diyl) dibenzoic acid, DMA=N, N-dimethylacetamide) have been synth-esized and identified by Infrared spectroscopy (IR), elemental analysis, thermogravimetric analysis (TG) and single crystal X-ray diffraction. Structural analyses show that 1 and 2 are isostructural, and display a 3D (3, 6)-connected net with the point symbol (42·6)·(44·62·88·10). Besides, the UV-Vis absorption spectra in solid state, the magnetic properties and catalytic activity of 1~2 were investigated. Between the proximal Co(Ⅱ)/Ni(Ⅱ) ions, the variable-temperature (2~300 K) magnetic susceptibilities of 1~2 display antiferromagnetic coupling. Furthermore, 1 have been verified to be effectual catalysts for the oxidation of arylacycloalkanes in aqueous medium.
-
0. Introduction
Complexes as one of the most attractively versatile platforms for achieving long-range organization and order as well as exploitable property, has been attractive in different areas during the past decade. Due to the adjustable flexibility and connectivity information of ligands, the judicious selection of organic ligands to coordinate with suitable metal centers is a key factor for the modulation of the structure and property of complex. Among this, a variety of complexes have been prepared under hydrothermal conditions by using unsymmetrical N-heterocyclic ligands and aromatic polycarboxylate ligands[1-4], which were favorable for the potential applications in gas adsorption, fluorescence sensing, catalysis and photocatalysis, and molecular switch[5-9]. Bifunctional pridine-carboxylate ligands possess N- and O- potential electron-donating centers which can exhibit diverse coordination modes with metal centers and various structures[10-13]. Notably, carboxylate groups on the ligands may facilitate the formation of discrete multinuclear clusters and infinite building blocks by (M-COO-M) linkage, such as Co(Ⅱ)/Ni(Ⅱ) complexes[14]. It is a crucial way to impart cluster-based complexes with expected magnetic properties and catalysts.
From the reported references, the study of complexes-based heterogeneous catalysts is concen-trated on the relation between the structures of hetero-geneous catalysts and the size of reaction substrates[15-20]. The influence of central metals to catalytic behaviors has not been systematically researched and discu- ssed[21-24]. It has been observed that the catalytic perfor-mance of complexes-based heterogeneous catalysts is strongly dependent on the metal entities[25]. To gain a deep understanding of the relationships between the metal ions and the catalytic behaviors, synthesis and research of isostructural complexes can exclude the influence of ligands and geometries.
Herein, 3, 3′-(pyridine-3, 5-diyl) dibenzoic acid (H2pddb) was selected as the organic linker with abundant N-donor sites and two carboxyl groups, which might be partially or completely deprotonated. Through the coordination reactions of Co(NO3)2·6H2O or Ni(NO3)2·6H2O with H2pddb, two isostructural compl-exes {[Co2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n (1) and {[Ni2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n (2) were obtained, which were identified by elemental analyses, IR, TG, single-crystal X-ray diffractions. In addition, for these complexes, the solid state absorption spectra, magnetic properties and catalytic activity have been investigated.
1. Experimental
1.1 General information and materials
3, 3′-(pyridine-3, 5-diyl) dibenzoic acid, other reagents and solvents employed were of AR grade from commercial sources and used as received. IR data were recorded on a BRUKER TENSOR 27 spectrophotometer with KBr pellets from 400 to 4 000 cm-1. Elemental analyses (C, H, and N) were carried out on a FLASH EA 1112 elemental analyzer. Powder X-ray diffraction (PXRD) patterns were recorded on a PANalytical X′Pert PRO diffractometer with Cu Kα (λ=0.154 18 nm) radiation at room temperature, using an operating tube voltage of 40 kV and tube current of 40 mA in a 2θ range between 5° and 50°. UV-Vis absorption spectra were obtained from Hewlett Packard 8453 UV-Vis Spectrophotometer. Dynamic magnetization experiments of polycrystalline samples were carried out with a SQUID MPMS XL-7 instrument at H=1 000 Oe over a temperature range of 2~300 K. NMR spectra were recorded on 400 MHz Bruker Avance-400 spectrometer and the chemical shift was reported relative to the signals of an internal standard of TMS (δ=0.0).
1.2 Synthesis of complexes
1.2.1 Synthesis of {[Co2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n (1)
A mixture of 3, 3′-(pyridine-3, 5-diyl) dibenzoic acid (0.009 6 g, 0.03 mmol), Co(NO3)2·6H2O (0.023 3 g, 0.08 mmol), H2O (3 mL), and DMA (3 mL) was poured into a vial of PTFE gasket (10 mL), then the vessel was sealed and heated to 100 ℃ for 3 days. The vial was cooled to room temperature at 5 ℃·h-1. Crystals of 1 suitable for X-ray analysis were collected. Anal. Calcd. for C46H56Co2N4O18(%): C, 51.55; H, 5.23; N, 5.23. Found(%):C, 50.98; H, 5.36; N, 5.02. IR (KBr, cm-1): 3 431 (w), 3 126 (w), 2 991 (w), 1 703 (w), 1 665 (m), 1 564 (s), 1 432 (m), 1 321 (s), 1 264 (w), 1 108 (w), 982 (w), 755 (m), 557 (w).
1.2.2 Synthesis of {[Ni2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n (2)
A mixture of 3, 3′-(pyridine-3, 5-diyl) dibenzoic acid (0.009 7 g, 0.03 mmol), Ni(NO3)2·6H2O (0.023 2 g, 0.08 mmol), H2O (3 mL), and DMA (3 mL) was poured into a vial of PTFE gasket (10 mL), then the vessel was sealed and heated to 100 ℃ for 3 days. The autoclave was cooled to room temperature at 5 ℃·h-1. Crystals of 2 suitable for X-ray analysis were collected. Anal. Calcd. for C46H56N4Ni2O18(%):C, 51.57; H, 5.23; N, 5.23. Found(%): C, 51.13; H, 5.46; N, 5.39. IR (KBr, cm-1): 3 418 (w), 3 024 (w), 2 889 (w), 1 698 (w), 1 612 (m), 1 544 (s), 1 419 (m), 1 319 (s), 1 255 (w), 1 106 (w), 998 (w), 769 (m), 541 (w).
1.3 Typical procedure for C-H bond activation of arylacycloalkane to ketones
A mixture of arylacycloalkane (1 mmol), tert-butyl hydroperoxide (TBHP) (1.5 mmol), and catalyst (0.05 mmol) in H2O (2 mL) was ultrasound at room temperature for 4 h in the air. After reaction finished, catalyst was recovered by centrifugation. The residue was extracted with ethyl acetate and H2O. The organic phases were combined, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel with petroleum ether/ethyl acetate (5:1, V/V) as eluent to afford product. The 1H NMR data of pure products were consistent with previous literature report (Supporting information)[26-28].
1.4 Single-crystal structure determination
The crystallographic data were collected on a Bruker D8 VENTURE diffractometer with Mo Kα radiation (λ=0.071 073 nm) at 298 K. The integration of the diffraction data and the intensity corrections were performed using the SAINT program[29]. Semiem-pirical absorption correction was performed using SADABS program[30]. The structures were solved by direct methods and refined with a full matrix least-squares technique based on F2 with the SHELXL-2016 crystallographic software package[31]. The hydrogen atoms except for those of water molecules were generated geometrically and refined isotropically using the riding model. Crystallographic data and structure processing parameters are summarized in Table 1.
Table 1

 下载:
导出CSV
下载:
导出CSV
Complex 1 2 Formula C46H56Co2N4O18 C46H56N4Ni2O18 Formula weight 1 070.81 1 070.36 Crystal system Monoclinic Monoclinic Space group C2/c C2/c a / nm 2.009 7(5) 1.997 8(8) b / nm 1.100 2(4) 1.094 5(3) c / nm 2.212 3(6) 2.197 0(5) β / (°) 92.750(7) 93.146(10) V / nm3 4.886(3) 4.797(3) Z 4 4 Dc / (g·cm-3) 1.456 1.482 μ / mm-1 0.757 0.865 2θ range / (°) 4.222~55.054 4.244~55.072 F(000) 2 232.0 2 240.0 Data, restraint, parameter 5 586, 0, 327 5 507, 0, 324 Reflection collected 38 980 37 799 Goodness of fit on F2 1.057 1.032 Final R indices [I > 2σ(I)] R1=0.041 5, wR2=0.113 9 R1=0.041 7, wR2=0.108 7 Final R indices (all data) R1=0.048 9, wR2=0.118 9 R1=0.057 4, wR2=0.116 9 Largest diff. peak and hole / (e·nm-3) 730, -600 560, -610 CCDC: 1952526, 1; 1952527, 2.
2. Results and discussion
2.1 Description of the crystal structures
2.1.1 Crystal structure of {[Co2(pddb)2(μ2-H2O)(H2O)2]·2DMA·5H2O}n (1)
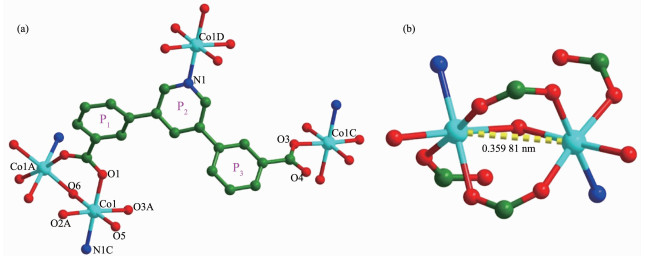
The X-ray crystallographic analysis reveals that complex 1 crystallizes in the monoclinic space group C2/c. The asymmetric unit of 1 consists of one Co(Ⅱ) ion, one pddb2- ligand, one coordinated water molecule, one half μ2-H2O, two and half lattice-water molecules, and one free DMA molecules. As shown in Fig. 1(a), each Co(Ⅱ) ion is six-coordinated with a regular octah-edron geometry, which is formed by three oxygen atoms (Co1-O: 0.207 05(16), 0.209 37(16) and 0.210 57(16) nm) from different pddb2- ligands, one nitrogen atom (Co1-N: 0.217 61(18) nm) from one pddb2- ligand, one oxygen atoms (Co1-O: 0.214 09(18) nm) from coordinated water molecule and one μ2-H2O (Co1-O: 0.216 03(13) nm).
Figure 1
 Figure 1. a) Coordination environments of the Co(Ⅱ) ions in 1; (b) [Co2(CO2)(μ2-H2O)] SBUs in 1
Figure 1. a) Coordination environments of the Co(Ⅱ) ions in 1; (b) [Co2(CO2)(μ2-H2O)] SBUs in 1Hydrogen atoms and free solvent molecules are omitted for clarity; Symmetry codes: A: 1-x, +y, 1/2-z; B: 3/2-x, 3/2-y, -z; C:-1/2+x, 1/2+y, +z; D: 1/2+x, -1/2+y, +z
Two Co(Ⅱ) ions are connected together by one μ2-H2O and two carboxylate group from pddb2- to form a second building unit (SBU) of [Co2(CO2)(μ2-H2O)] with a Co…Co distance of 0.359 81 nm (Fig. 1(b)). The coor-dination mode of the pddb2- ligand can be described as μ3-η1:η1:η1 (Fig. 1(a)). The dihedral angle between the plane P1 and plane P2 is 36.324°, and the dihedral angle between the plane P2 and plane P3 is 41.706°. A better insight into the present 3D framework can be accessed by the topological method. As shown in Fig. 2(a), each [Co2(CO2)(μ2-H2O)] SBUs coordinates six different pddb2- ligands and can be simplified as a 6-connected node, and one pddb2- links three [Co2(CO2) (μ2-H2O)] SBUs, serving as a 3-connected node. There-fore, the structure of complex 1 is a binodal 3, 6-connected 3D framework with a point symbol of (42·6)·(44·62·88·10), as depicted in Fig. 2(b). Finally, 3D supramolecular structure is packed through twelve sorts of hydrogen bonding (classical hydrogen bonding: O-H…O and C-H…O), as shown in Fig. 3.
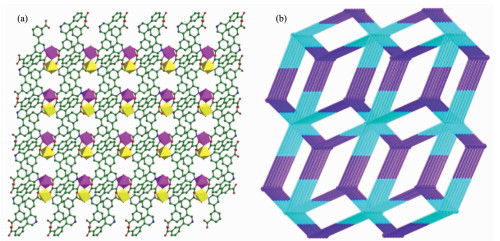
Figure 2
 Figure 2. (a) 3D structure of 1 viewed along a axis; (b) Topological structure of (3, 6)-connected with 1 nodal net
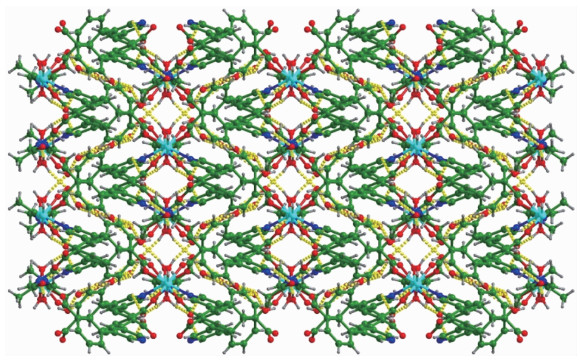
Figure 2. (a) 3D structure of 1 viewed along a axis; (b) Topological structure of (3, 6)-connected with 1 nodal netFigure 3
 Figure 3. Perspective view of the supramolecular framework through twelve kinds of hydrogen-bonding interactions (dotted line)
Figure 3. Perspective view of the supramolecular framework through twelve kinds of hydrogen-bonding interactions (dotted line)2.1.2 Crystal structure of {[Ni2(pddb)2(μ2-H2O)(H2O)2] ·2DMA·5H2O}n (2)
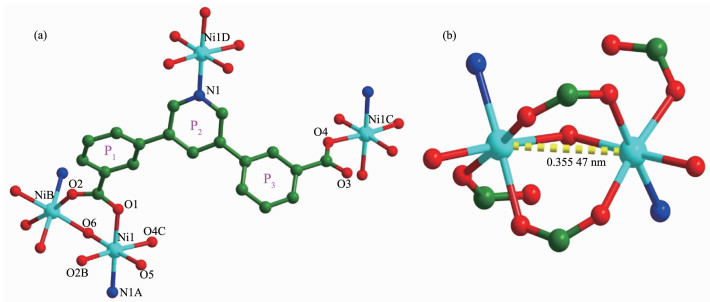
The single-crystal X-ray diffraction study revealed that 1 and 2 are isostructural and H2pddb ligands show same coordination mode. Co(Ⅱ) ions in 1 are replaced by Ni(Ⅱ) ions, and there are one free DMA molecule and 2.5 lattice-water molecules in 2 (Fig. 4(a)). Comparing with that of complex 1, the Ni…Ni distance in the dinuclear unit [Ni2(COO)2(μ2-H2O)] is 0.355 47 nm (Fig. 4(b)). The dihedral angle between the planes P1 and P2 is 35.824°, and the dihedral angle between the planes P2 and P3 is 41.884°. In the crystal structure of 2, through intramolecular and intermolecular O-H…O and C-H…O hydrogen bonds, adjacent molecules are linked together into 3D supramolecular networks.
Figure 4
 Figure 4. (a) Coordination environments of the Ni(Ⅱ) ions in 2; (b) [Ni2(CO2)(μ2-H2O)] SBUs in 2
Figure 4. (a) Coordination environments of the Ni(Ⅱ) ions in 2; (b) [Ni2(CO2)(μ2-H2O)] SBUs in 2Symmetry codes: A: 1/2+x, 1/2+y, z; B:-x, y, 1/2-z; C:-1/2-x, 5/2-y, 1-z; D:-1/2+x, -1/2+y, z
2.2 Phase purity and thermogravimetric analysis
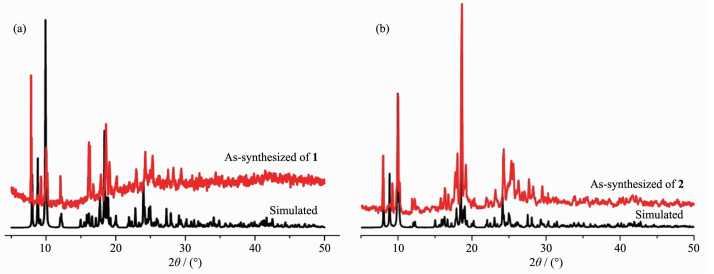
To check the phase purity of complexes 1~2, X-ray powder diffraction analyses were checked at room temperature. As shown in Fig. 5, the experimental patterns match well with the simulated ones, which manifests the high crystallinity and good phase purities of the samples.
Figure 5
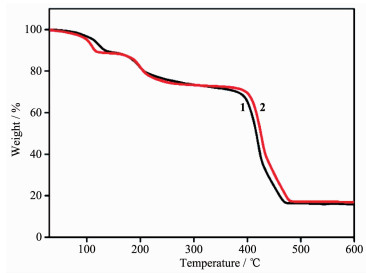
In addition to the phase purity, the thermal properties and the change of crystal forms of 1~2 were investigated by using thermogravimetric analysis (TGA) (Fig. 6). For complex 1, the first weight loss was from 30 to 125 ℃ (Obsd. 8.36%, Calcd. 8.41%), corresponding to the removal of the lattice H2O molecules. The second weight loss of 19.88% (Calcd. 19.64%) corresponds to the loss of free DMA molecule and coordinated water molecules. The removal of μ2-H2O and organic ligands occurred within the range of 350~480 ℃, finally leading to the formation of the stoichiometric amount of CoO as a residue. The TG curve for 2 also showed an obvious weight loss between 60 and 350 ℃, which can be ascribed to the removal of lattice water molecules, free DMA molecule and coordinated water molecules (Obsd. 27.41%, Calcd. 28.08%). The removal of organic ligands and μ2-H2O occurred within the range of 390~490 ℃. The remaining weight corresponds to the formation of NiO.
Figure 6
2.3 UV-Vis spectra analyses
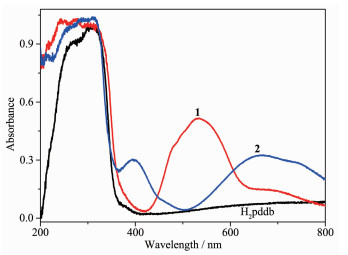
The solid state absorption spectra of free ligand H2pddb, 1 and 2 at room temperature were investigated. The electronic absorption spectrum of free ligand H2pddb exhibited two absorption peaks at approxi-mately 263 and 310 nm. They can be assigned to the π-π* transition of benzene rings and the intraligand π-π* transition of the C=N group[32]. The absorption spectra of complexes 1 and 2 were obviously different from those of H2pddb (Fig. 7). In 1, there are several intense absorption bands less than 400 nm and one intermediate absorption band around 535 nm. The absorption band from 200 to 400 nm should be considered as π-π* transitions of the H2pddb[33]. Another absorption band from 400 to 700 nm can be ascribed to the spin-allowed d-d electronic transitions of the d7 (Co2+) cation[34]. As depicted in Fig. 7, the spectra of complex 2 display three wide absorption bands in a range of 200~350 nm, 350~500 nm and 500~800 nm, respectively. The band at 394 and 670 nm are assignable to metal-to-ligand charge transfer (MLCT) transitions (3A2g-3T1g), indicating octahedral geometry[35].
Figure 7
2.4 Magnetic property
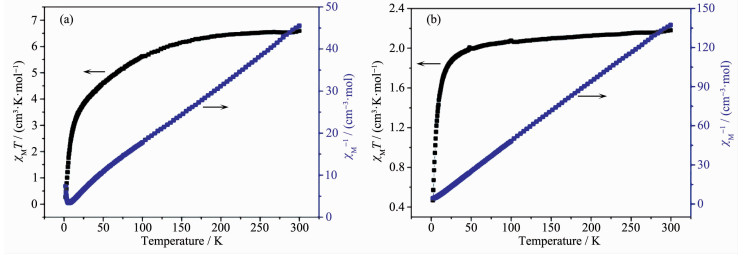
The magnetic properties of complexes 1 and 2 were investigated over the temperature range of 2~300 K at an applied field of 1 000 Oe. As shown in Fig. 8(a), the χMT value of 1 at 300 K was 6.59 cm3·mol-1·K, which is larger than the expected value of 3.75 cm3·mol-1·K for one isolated Co(Ⅱ) (S=3/2, g=2) ion[8]. As the temperature decreased, the value of χMT continuously decreased and reached a minimum value of 0.27 cm3·mol-1·K at 2 K. The data above 10~300 K fitted the Curie-Weiss law well with Curie constant C=7.08 cm3·mol-1·K and Weiss constant θ=-23.15 K (Fig. 8(a)). The large negative θ value indicates dominant antiferromagnetic coupling[36-38]. For the Ni(Ⅱ) complex 2, the χMT value at 300 K was 2.18 cm3·mol-1·K, which is in agreement with the spin-only value of 2.0 cm3·mol-1·K for one isolated Ni(Ⅱ) (S=1/2, g=2.0) ion. Upon lowering the temperature, the χMT value decreased slowly until about 50 K, then decreased quickly to a minimum value of 0.47 cm3·mol-1·K at 2 K[8]. The data above 50~300 K fitted the Curie-Weiss law well with Curie constant C=2.19 cm3·mol-1·K and Weiss constant θ=-5.12 K (Fig. 8(b)). The negative θ value and the decrease of χMT value suggest the dominant antiferro-magnetic interactions above 50 K[36-38]. In 1 and 2, two metal ions are connected by one μ2-H2O and two carboxylate group to form a magnetic exchange pathway of {…M-O-C-O-M…}.
Figure 8
 Figure 8. Temperature dependence of the χMT values for 1 (a) and 2 (b) at 1 000 Oe dc magnetic field
Figure 8. Temperature dependence of the χMT values for 1 (a) and 2 (b) at 1 000 Oe dc magnetic field2.5 Catalytic capacity
As is known to all, the carbonyl compounds represent important applications in dyes, drugs and other fine chemicals. Inspired by unique advantages of metal organic frameworks materials[39], we decided to explore the catalytic performance of Co(Ⅱ)/Ni(Ⅱ) complexes in the oxidation of arylacycloalkane, and investigated the influence of central metals to catalytic behaviors. To expand the scope of complexes for oxidation of C-H bond, four substrates, including cyclanes and heterocyclic alkanes, were subjected to ketone under the optimized conditions (Table 2). It is obvious that the catalyst 1 behaved excellent catalytic performance, and the reason for this is Ni2+ is difficult to be completely oxidized to Ni3+ during the catalytic process. Proposed mechanism for the oxidation reaction indicated that there was a reversible oxidation/reduction transformation of central metal ions[28].
Table 2
Table 2. Oxidation of arylacycloalkane catalyzed by Co(Ⅱ)/Ni(Ⅱ) complexes下载:
导出CSV

Entry Catalyst Substrate Product Yield/% 1 1 

84 2 2 — 3 1 

70 4 2 — 5 1 

72 6 2 — 7 1 

85 8 2 — 3. Conclusions
Two 3D complexes have been obtained by the coordination reactions of Co(Ⅱ)/Ni(Ⅱ) salts with 3, 3′-(pyridine-3, 5-diyl) dibenzoic acid (H2pddb). Complexes 1 and 2 are isostructural, and H2pddb ligands show same coordination modes. Magnetic studies for complexes 1 and 2 show an antiferromagnetic coupling between the adjacent Co(Ⅱ)/Ni(Ⅱ) centers. Furthermore, the catalyst 1 behaved excellent catalytic performance for the oxidation of arylacycloalkanes in aqueous medium.
Supporting information is available at http://www.wjhxxb.cn
-
-
[1]
Zhang J, Li B D, Wu X J, et al. J. Mol. Struct., 2010, 984:276-281 doi: 10.1016/j.molstruc.2010.09.040
-
[2]
Shen P P, Ren N, Zhang J J, et al. J. Therm. Anal. Calorim., 2019, 135:2687-2695 doi: 10.1007/s10973-018-7265-0
-
[3]
Fan Y R, Li H B, Jia Z Y, et al. Inorg. Chem. Commun., 2019, 102:229-232 doi: 10.1016/j.inoche.2019.02.037
-
[4]
Xing Y B, Liu Y Q, Meng B F, et al. J. Chem. Crystallogr., 2019:1-8
-
[5]
Wu Y P, Xu G W, Dong W W, et al. Inorg. Chem., 2017, 56:1402-1411 doi: 10.1021/acs.inorgchem.6b02476
-
[6]
Ma K, Zhao Y N, Han X, et al. Cryst. Growth Des., 2018, 18:7419-7425 doi: 10.1021/acs.cgd.8b01113
-
[7]
Maity D K, Dey A, Ghosh S, et al. Inorg. Chem., 2018, 57:251-263 doi: 10.1021/acs.inorgchem.7b02435
-
[8]
Shao Z C, Han X, Liu Y Y, et al. Dalton Trans., 2019, 48:6191-6197 doi: 10.1039/C9DT00968J
-
[9]
Sun Y Y, Wang F, Zhang J. Inorg. Chem., 2019, 58:4076-4079 doi: 10.1021/acs.inorgchem.9b00261
-
[10]
Zhao B, Li H J, Jia Y Y, et al. Polyhedron, 2014, 68:138-143 doi: 10.1016/j.poly.2013.10.022
-
[11]
王桂仙, 曹可利, 陈飞燕, 等.无机化学学报, 2016, 32: 43-48 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20160106&flag=1WANG Gui-Xian, CAO Ke-Li, CHEN Fei-Yan, et al. Chinese J. Inorg. Chem., 2016, 32: 43-48 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20160106&flag=1
-
[12]
林建军, 刘珍, 吕灵芝, 等.无机化学学报, 2018, 34: 230-236 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180202&flag=1LIN Jian-Jun, LIU Zhen, LÜ Ling-Zhi, et al. Chinese J. Inorg. Chem., 2018, 34: 230-236 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180202&flag=1
-
[13]
Zhao L, Zhang J, Wang J, et al. J. Solid State Chem., 2018, 268:1-8 doi: 10.1016/j.jssc.2018.08.028
-
[14]
Liu L, Huang C, Xue X, et al. Cryst. Growth Des., 2015, 15:4507-4517 doi: 10.1021/acs.cgd.5b00757
-
[15]
Corma A, García H, Xamena F X L I. Chem. Rev., 2010, 110:4606-4655 doi: 10.1021/cr9003924
-
[16]
Yoon M, Srirambalaji R, Kim K. Chem. Rev., 2012, 112:1196-1231 doi: 10.1021/cr2003147
-
[17]
Zhao M, Ou S, Wu C D. Acc. Chem. Res., 2014, 47:1199-1207 doi: 10.1021/ar400265x
-
[18]
Liu J, Chen L, Cui H, et al. Chem. Soc. Rev., 2014, 43:6011-6061 doi: 10.1039/C4CS00094C
-
[19]
Dhakshinamoorthy A, Asiri A M, Garcia H. Chem. Soc. Rev., 2015, 44:1922-1947 doi: 10.1039/C4CS00254G
-
[20]
Zhu Q L, Xu Q. Chem. Soc. Rev., 2014, 43:5468-5512 doi: 10.1039/C3CS60472A
-
[21]
Wang H R, Meng W, Wu J, et al. Coord. Chem. Rev., 2016, 307:130-146 doi: 10.1016/j.ccr.2015.05.009
-
[22]
Park S S, Hontz E R, Sun L, et al. J. Am. Chem. Soc., 2015, 137:1774-1777 doi: 10.1021/ja512437u
-
[23]
Chughtai A H, Ahmad N, Younus A Y, et al. Chem. Soc. Rev., 2015, 44:6804-6849 doi: 10.1039/C4CS00395K
-
[24]
Yao H F, Yang Y, Liu H, et al. J. Mol. Catal. A:Chem., 2014, 394:57-65 doi: 10.1016/j.molcata.2014.06.040
-
[25]
Guo L, Huang C, Liu L, et al. Cryst. Growth Des., 2016, 16:4926-4933 doi: 10.1021/acs.cgd.6b00494
-
[26]
Shaabani A, Farhangi E, Rahmati A. Appl. Catal. A, 2008, 338:14-19 doi: 10.1016/j.apcata.2007.12.014
-
[27]
Hossain M M, Shyu S. Tetrahedron, 2016, 72:4252-4257 doi: 10.1016/j.tet.2016.05.066
-
[28]
Gao K, Huang C, Yang Y, et al. Cryst. Growth Des., 2019, 19:976-982 doi: 10.1021/acs.cgd.8b01527
-
[29]
SAINT, Bruker AXS, Inc., Madison, WI, 2001.
-
[30]
Sheldrick G M. SADABS, University of Göttingen, Germany, 2003.
-
[31]
Sheldrick G M. Acta Crystallogr. Sect. A:Found. Crystallogr., 2008, A64:112-122
-
[32]
Liu P P, Wang C Y, Zhang M, et al. Polyhedron, 2017, 129:133-140 doi: 10.1016/j.poly.2017.03.019
-
[33]
Zhou S B, Wang X F, Du C C, et al. CrystEngComm, 2017, 19:3124-3137 doi: 10.1039/C7CE00394C
-
[34]
Yan W, Han L J, Jia H L, et al. Inorg. Chem., 2016, 55:8816-8821 doi: 10.1021/acs.inorgchem.6b01328
-
[35]
Fan C, Zong Z, Zhang X, et al. CrystEngComm, 2018, 20:4973-4988 doi: 10.1039/C8CE00868J
-
[36]
Li D S, Zhao J, Wu Y P, et al. Inorg. Chem., 2013, 52:8091-8098 doi: 10.1021/ic4007718
-
[37]
Zeng M H, Zhou Y L, Wu M C, et al. Inorg. Chem., 2010, 49:6436-6442 doi: 10.1021/ic100021f
-
[38]
Qin J, Jia Y, Li H, et al. Inorg. Chem., 2014, 53:685-687 doi: 10.1021/ic402598p
-
[39]
Wang X, Liu M, Wang Y, et al. Inorg. Chem., 2017, 56:13329-13336 doi: 10.1021/acs.inorgchem.7b02106
-
[1]
-
Figure 1 a) Coordination environments of the Co(Ⅱ) ions in 1; (b) [Co2(CO2)(μ2-H2O)] SBUs in 1
Hydrogen atoms and free solvent molecules are omitted for clarity; Symmetry codes: A: 1-x, +y, 1/2-z; B: 3/2-x, 3/2-y, -z; C:-1/2+x, 1/2+y, +z; D: 1/2+x, -1/2+y, +z
Figure 2 (a) 3D structure of 1 viewed along a axis; (b) Topological structure of (3, 6)-connected with 1 nodal net
Figure 3 Perspective view of the supramolecular framework through twelve kinds of hydrogen-bonding interactions (dotted line)
Figure 4 (a) Coordination environments of the Ni(Ⅱ) ions in 2; (b) [Ni2(CO2)(μ2-H2O)] SBUs in 2
Symmetry codes: A: 1/2+x, 1/2+y, z; B:-x, y, 1/2-z; C:-1/2-x, 5/2-y, 1-z; D:-1/2+x, -1/2+y, z
Figure 8 Temperature dependence of the χMT values for 1 (a) and 2 (b) at 1 000 Oe dc magnetic field
Table 1. Crystal data and structure refinement data of complexes 1~2
Complex 1 2 Formula C46H56Co2N4O18 C46H56N4Ni2O18 Formula weight 1 070.81 1 070.36 Crystal system Monoclinic Monoclinic Space group C2/c C2/c a / nm 2.009 7(5) 1.997 8(8) b / nm 1.100 2(4) 1.094 5(3) c / nm 2.212 3(6) 2.197 0(5) β / (°) 92.750(7) 93.146(10) V / nm3 4.886(3) 4.797(3) Z 4 4 Dc / (g·cm-3) 1.456 1.482 μ / mm-1 0.757 0.865 2θ range / (°) 4.222~55.054 4.244~55.072 F(000) 2 232.0 2 240.0 Data, restraint, parameter 5 586, 0, 327 5 507, 0, 324 Reflection collected 38 980 37 799 Goodness of fit on F2 1.057 1.032 Final R indices [I > 2σ(I)] R1=0.041 5, wR2=0.113 9 R1=0.041 7, wR2=0.108 7 Final R indices (all data) R1=0.048 9, wR2=0.118 9 R1=0.057 4, wR2=0.116 9 Largest diff. peak and hole / (e·nm-3) 730, -600 560, -610  下载: 导出CSV
下载: 导出CSV
Table 2. Oxidation of arylacycloalkane catalyzed by Co(Ⅱ)/Ni(Ⅱ) complexes
Entry Catalyst Substrate Product Yield/% 1 1 84 2 2 — 3 1 70 4 2 — 5 1 72 6 2 — 7 1 85 8 2 —
下载: 导出CSV
-









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 2
- 文章访问数: 1627
- HTML全文浏览量: 87
