Citation:
LIU Jun, ZHOU Quan, XIE Jia-Qi, LI Rong. Preparation and High Electrocatalytic Activity for the Oxidation of Ethanol of PdAu Alloy Foam Films[J]. Chinese Journal of Inorganic Chemistry,
2020, 36(1): 21-30.
doi:
10.11862/CJIC.2019.195
Received Date:
14 May 2019 Revised Date:
25 June 2019 Available Online:
10 January 2020
Abstract:
Three-dimensional (3D) porous Au-doped PdAu alloy foam films were obtained by using a hydrogen bubble dynamic template electrodeposition method. The morphology and structure features of 3D porous Au-doped Pd alloy foam films were characterized by field-emission scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). Due to the special porous structures and the electronic effects, the Au-doped PdAu alloy foam films showed high electrocatalytic activity toward the electrooxidation of ethanol in alkaline media as compared to pure porous Pd film.
Monometallic Pd nano-materials as efficient catalysts have a widespread use in diverse fields of applications[1-4]. Recent investigations have suggested that introduction of a second metal into the Pd matrix can give rise to the added advantages of ligand effect, strain effect and ensemble effect resulting in improved reactivity of the Pd-based catalysts[5-8]. On the basis of this fact, a number of strategies have been proposed for improving the performance of an electrocatalyst activity that involved alloying Pd with other transition metals such as PdAg[5], PdAu[6], PdPt[7] and PdCo[8]. It is generally accepted that the enhancement of the activity of these metals compared to pure Pd is due to the so-called bifunctional and/or electronic effect[5-8]. It has been well-known that the incorporation of Au into Pd catalysts can also improve catalytic activity and selectivity as well as the resistance to poisoning[6, 9-11]. Consequently, much research efforts have been paid on controllably fabricating the PdAu bimetallic structures and deliberately controlling their structures at nanoscale[6, 9-11]. Various Pd-Au bimetallic architec-tures have been synthesized, and their catalytic activities have been evaluated. But a number of reports have focused on the preparation and characterization of Pd-Au bimetallic nanoparticles[12-14], the synthesis of Pd-Au alloy catalysts with unconventional shapes and controlled atomic distribution remains relatively unexplored.
As we known, 3D porous nanomaterials have been explored for a new generation of advanced devices such as sensors[15], batteries[16] and fuel cells[17] because of their great potential for rapid electro-chemical reactions arising from the extremely large specific surface areas for charge and mass (gas) transport. To data, templating[18-19] and dealloying[20-22] are two most popular methods while they involved multisteps to get the ultimate porous structures. Attempts have been made to overcome these drawbacks by synthesize 3D macroporous lms in a complicated way. Currently, a novel and simple hydrogen bubble dynamic template have been developed to produce self-supported 3D porous nanometals, such as Au[23], Ni[24], PdPt[25], AuPt[26], Cu[27]. The advantage of this approach is that it is rapid, clean and does not result in any contamination of the metal surface with organic species.
Most recently, we synthesized 3D noble alloy foam films of Au utilizing the hydrogen bubble dynamic template method in H2SO4 solutions containing low concentration of precursors[23]. However, to the best of the authors knowledge, it has not been applied to the direct formation of highly porous PdAu alloy. In this paper, we have successfully sculptured a variety of three-dimensional porous Au-doped PdAu alloy foam films (3D PPAFs) with open interconnected macrop-orous walls and nanoparticles using hydrogen bubbles as the dynamic template. Furthermore, relative molar ratios between Pd and Au at the porous structures could be controlled by adjusting the molar ratios between metal precursors in the feeding solutions. Using this simple approach, we routinely produced PdAu alloyed nanostuctures with high surface areas and the particularly catalytic activity for the methanol oxidation compared with that of three-dimensional porous Pd foam films (3D PPFs). It was found that the intrinsic activity of the alloyed catalysts for ethanol oxidation increased with the increase of the Pd:Au ratio until it reached a maximum for 100:3 in feeding solution. This synthesis also provides a convenient and environmentally benign route to production because it does not require high temperature, organic solvent, surfactant and time saving.
1.
Experimental
1.1
Reagents
PdCl2 and HAuCl4·4H2O were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Sulfuric acid, sodium hydroxide and methanol were purchased from the Factory of Hunan Normal University. All the chemicals were of analytical grade and were used as received. Milli-Q water with a resistivity of greater than 18.3 MΩ·cm was used in the preparation of aqueous solutions.
1.2
Electrodeposition of 3D porous PdAu, Pd and Au foam films
Electrochemical experiments were performed on a CHI 660C electrochemical workstation (Chenhua Instruments, Shanghai, China). A palladium disk (1 mm diameter, purity 99.99%), a platinum foil (geo-metric area 1 cm2), and a saturated mercurous sulfate electrode (SMSE) were employed as the working, counter, and reference electrodes, respectively. Prior to use, the working electrode was polished with 2 000 grit carbimet paper, followed by rinsing in millipore water under ultrasonic waves.
Then, the well-polished electrode was electro-chemically pretreated by cycling the potential between -0.72 and +0.70 V in 0.5 mol·L-1 H2SO4 at a scan rate of 100 mV·s-1 until a stable voltammogram was obtained. Then the palladium electrode was rinsed with millipore water. 3D porous PdAu lms were fabricated by electrodeposition under constant potential of -4 V for 200 s. We denoted the deposited foam films as Pd100Au1, Pd100Au3, and Pd100Au5, respectively, according to the mole ratio of PdCl2/HAuCl4 in the feeding solutions: 2 mmol·L-1 PdCl2+x mmol·L-1 HAuCl4+2 mol·L-1 H2SO4 (x=0.02, 0.06 and 0.10). For comparison, monometallic 3D porous Pd and Au films were deposited in the same way by substituting the mixed precursors with 2 mmol·L-1 PdCl2 or 2 mmol·L-1 HAuCl4, respectively.
The deposited PdAu alloy and respective pure metal catalysts were firstly treated by cyclic voltam-metry between -0.72 and 0.70 V at 100 mV·s-1 in 0.5 mol·L-1 H2SO4 solution until a steady cyclic voltammo-gram (CV) was obtained (6 circles). The current densities of PdAu alloy catalysts for ethanol oxidation were normalized to the total electrochemical active surface areas (ECSAs).
1.3
Electrooxidation of ethanol
The electrocatalytic activity toward electrooxida-tion of ethanol was evaluated in solutions of 0.5 mol·L-1 NaOH+0.5 mol·L-1 CH3CH2OH. And the CVs were recorded in the potential ranges between -1.6 and 0.2 V for the oxidation of ethanol, at a scan rate of 20 mV·s-1. The current density-time (j-t) curves were measured at a fixed potential for 2 000 s. The current densities of PdAu and Pd foam catalysts for electrocatalytic oxidation of ethanol were normalized to the total EASAs, which were calculated using 424 μC·cm-2 for the reduction of Pd oxide monolayer[28].
1.4
Instrumentals
Scanning electron microscopy (SEM) images and energy-dispersive X-ray spectroscopy (EDX) data were obtained with a JEOL JSM-6360 scanning electron microscope operating at 25 kV after drying the samples. X-ray diffraction (XRD) analysis of the resulting products was carried out on a Dmax Rapid IIR diffractometer (Cu Kα radiation with λ=0.154 2 nm) at a scan rate of 2°·min-1 with 20°~120°, and the exposure time was 15 min at 40 kV and 40 mA. X-ray photoelectron spectroscopy (XPS) analysis was carried out on an ESCALAB MK Ⅱ X-ray photoelectron spectrometer with Al Kα X-ray radiation for excitation.
2.
Results and discussion
2.1
Characterizations of the electrodeposited foam films
The SEM images of the typical as-deposited porous Pd and Pd100Au3 foams were taken at different magnifications are shown in Fig. 1(a, c, e) and (b, d, f), respectively. Continuous network of Pd/PdAu was formed on the substrate (Fig. 1(a~d)) where the pore size increased from the bottom to the top of the sample. This was due to the coalescence of hydrogen bubbles that evolved from the surface. The foam walls were composed of hierarchical deposits, such a mocroporous structure was similar to porous Au films reported in recent literatures[23]. Interestingly, 3D PPFs and sample Pd100Au3 were similar in surface pore size (Fig. 1(c, d)). These results suggested that the low doping density of gold is little effect on the pore size of the foam. However, the morphology of the nano-clusters can be modified slightly by doping of Au (Fig. 1(e, f)). Before addition of Au, the foam walls of 3D porous Pd catalyst were composed of nanometer scale irregular Pd particles. The surface of bimetallic PdAu was tending to form dendrites which could enlarge surface area and might enhance catalytic active sites. Such an open porous structure can facilitate fast mass transfer of gas and liquid, while the extremely high area is good for electrocatalytic reactions[25-26].
Figure 1
Figure 1.
SEM images of as-prepared 3D PPF samples (a, c, e) and 3D porous Pd100Au3 films (b, d, f) by co-deposition along with H2 evolution at a constant potential of -4 V for 200 s taken with magnifications 200 (a, b), 3 000 (c, d) and 30 000 (e, f)
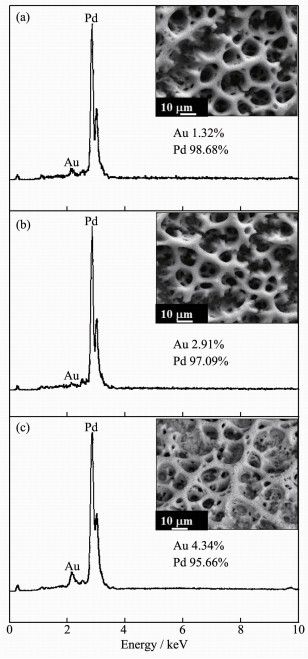
To examine chemical purity and composition of as-deposited bimetallic PdAu samples, EDX analysis were performed. Pd peaks at about 2.85 keV and Au signals at about 2.13 keV were observed. The corresponding EDX spectrum (Fig. 2) revealed that the atom ratio of Au in Pd100Au1, Pd100Au3, and Pd100Au5 samples were 1.32%, 2.91%, and 4.34%, respectively, in good agreement with stoichiometric PdAu within experimental error. Furthermore, the content of Au in feeding solution and EDX results are compared in Table 1. In short, the above analysis suggest that the compositions of bimetallic films are roughly consistent with those of the feeding solutions, as well as indicate that electrodeposition was controlled by the diffuse rate.
Figure 2
Figure 2.
EDX analysis of Pd100Au1 (a), Pd100Au3 (b) and Pd100Au5 (c)
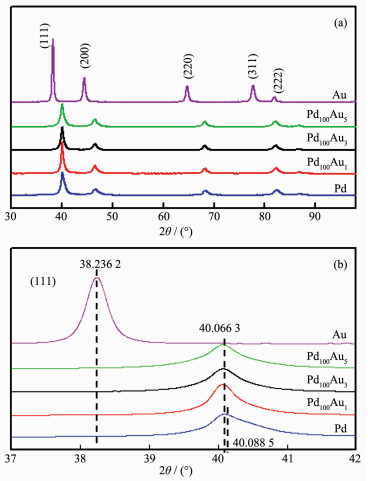
Further characterization by XRD was carried out, and Fig. 3 shows a typical XRD patterns of the 3D PPFs and PPAFs (Pd100Au1, Pd100Au3 and Pd100Au5). The peaks around 38.25°, 44.34°, 64.63°, 77.65°, 81.89°, 98.38°, 110.98°and 115.55° were assigned to Au (111), (200), (220), (311), (222), (400), (331) and (420) planes, respectively, according to PDF No.04-0784 of Au; and the peaks around 40.08°, 46.47°, 68.26°, 82.33°, 86.81°, 104.89° and 124.79° were assigned to Pd (111), (200), (220), (311), (222), (400) and (422) planes, respectively, according to PDF No.46-1043 of Pd.
Figure 3
Figure 3.
(a) XRD patterns and (b) the corresponding enlarged patterns of (111) diffraction peaks comparison
For the sake of comparison, the XRD patterns between the range of 39° and 42° of the samples were enlarged in Fig. 3b. Careful comparisons show that the peaks of Pd100Au5, Pd100Au3 and Pd100Au1 samples fall well between the (111) peaks of pure Pd and Au, suggesting that the PdAu porous films are, indeed, a single-phase alloy nanomaterial rather than a mixture of monometallic Pd and Au or a core-shell structure of Pd and Au. Similar results were obtained by our group in previous work[26]. In order to compare d-space parameters of each sample conveniently for readers, the refined d-space parameters of all samples are presented in Table 2. As we known, the reducer would affect the morphology and composition of bimetallic nanostructure because of the different reducing power. Such as, PdAu alloy nanodendrites were synthesized through mixing Na2PdCl4, HAuCl4, polyvinylpy and hydroquinone and heated at 50 ℃ for 15 min[29]. Meanwhile, PdAu-Au core-shell nanoparti-cles were synthesized using electrodeposition and displacement reaction[30]. On the basis of the reduction potentials of Au(Ⅲ)and Pd(Ⅱ)((AuCl4-/Au, 1.002 V vs SHE) and (PdCl42-/Pd, 0.59 V vs SHE)), in the present work, it can be assumed that the formation of the alloy structures should be co-deposited simultaneously of Pd and Au precursors on the surface of the Pd substrate because of the strong reducing power of tempestuously hydrogen evolution.
Table 2
Table 2.
Lattice spacings (d) of 3D porous PdAu alloys and pure Pd, pure Au sample
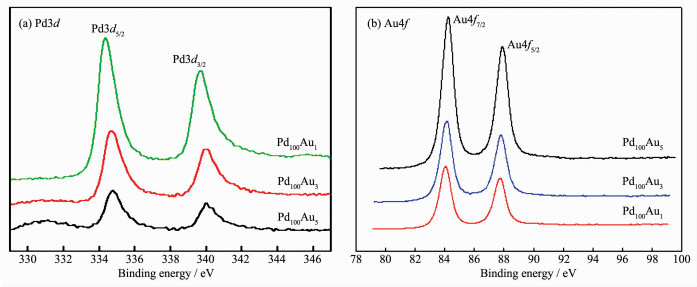
XPS characterizations were carried out to get surface composition of the electrodeposited alloy foam films. Fig. 4 presents the Pd3d and Au4f spectra from the 3D porous PdAu catalysts with different precursor atomic ratios. The binding energies (BEs) of the 3d signals for Pd and Au components in the deposited samples were summarized in Table 3. In Fig. 4a the BEs of 3d5/2 and 3d3/2 for metallic Pd decreased with the increase of Pd content. The BEs of Au4f7/2 and Au4f5/2 got lower with the increase of Au content in the PdAu foam films (Fig. 4b). These changes of BEs suggest that Au and Pd in the foams are atomically mixed with no phase separation, and they can be attributed to the electron effect and the geometric effect by the alloying. The electronic effect is a result of the d-band center shift of Pd and Au in the PdAu alloys. The geometric effect is defined in terms of the arrangement of surface atoms of a particular pattern required for being active to a given kind of reactant molecules. These are the main theoretical descriptions currently employed for the bimetallic electrocatalysts. Besides, the relative abundances at the surface of the PdAu foam films detected by XPS are also summarized in Table 3. The atomic percent of Pd in the samples of Pd100Au1, Pd100Au3, and Pd100Au5 obtained from the XPS were somewhat higher than that in the feeding solutions. The enrichment of Pd on the surface of the porous structures might be due to the stronger hydrogen adsorption on Pd than on Au at -4 V.
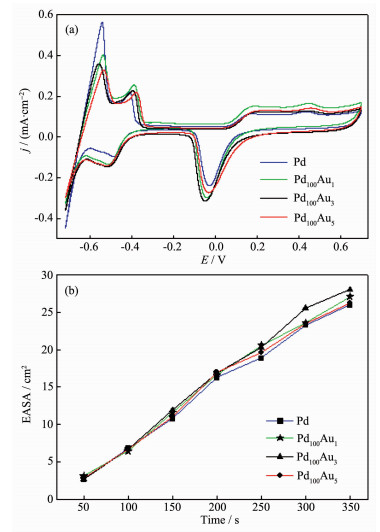
Electrochemical behaviors of such 3D PPF and PPAFs were investigated by CV in the potential range between -0.72 and -0.70 V in 0.5 mol·L-1 H2SO4 solutions. The CV curves after electrode stable are presented in Fig. 5a. The voltammetric features of the bimetallic PdAu electrodes were similar to the pure porous Pd film electrode (the blue line), herein, the adsorption and desorption of hydrogen was within the potential range of -0.72 to -0.35 V, the double-layer capacitance region was between -0.35 and 0.07 V, the formation of surface oxide at potentials was more positive than 0.07 V, and the reduction of the surface oxide is in the cathodic potential scan. As we known, Pd exhibit strong hydrogen storage ability which makes it is different from the other noble metal. It should be noticed that the peak current densities for hydrogen desorption (Hdes) of the PdAu bimeallic catalysts showed lower shift as the increasing of doping Au compared with the pure Pd catalysts, which might indicate different arrangement transformation of Au atoms in the Pd catalyst. However, there was no obvious difference of CVs between pure Pd and PdAu alloy for that the lower content of Au on the alloy surface was not sufficient to affect the CV character of Pd catalyst. This result was consistent with the SEM images in Fig. 1. Consequently, current densities of PdAu alloys could be normalized by the ECSA value of Pd. Herein, the deposited PdAu alloy catalysts were firstly treated by CV in 0.5 mol·L-1 H2SO4 solution until a steady CV was obtained in a short time, the influence from the dissolution of the Au could be negligible in such a short electrodeposition time (200 s) and in 6 circles CV. Next, we studied the relationship of the ECSAs and the electrodeposition time which is show in Fig. 5b. The ECSAs were estimated from the CVs in 0.5 mol·L-1 H2SO4 solution by integrating the reduction charge of palladium oxide monolayer. It is proved that the ECSAs of all catalysts enlarged with the electrodeposition time. Also, we observed that the thickness of the 3D PPFs and 3D PPAFs increased with the deposition time. Before Au doping, the foam walls of 3D porous Pd catalyst were composed of nanometer scale irregular Pd particles as already discussed. The morphology of electrodeposited foams could be modified slightly by doping ounce of Au, which made the alloy surface tend to form dendrite resulting enlarge surface area. For a further comparison, the experimental results of Fig. 5b showed that the ECSAs of the porous Pd and PdAu electrodes were in accordance with each other in the same deposition time. Apparently, it was relative to the subtle difference morphology between them which showed in Fig. 1. In other words, a small amount of Au doping had little effect on the surface area. Combined with the XPS characterization results, the enrichment of Pd on the surface of the porous foam film weakened the effect of Au on the surface area, which made the surface areas of each electrodes in accordance with each other.
Figure 5
Figure 5.
(a) CV curves of as-prepared 3D PPF and PPAFs electrodes (deposition time, 200 s; electrolyte, 0.5 mol·L-1 H2SO4 aqueous solution; sweep rate, 100 mV·s-1; 25 ℃); (b) Dependence of ECSAs on the electrodeposition time
In consider of the roughness surface large enough and economical of Pd precursor, in this study, 200 s was chosen as an appropriate treatment time for the construction of 3D PPFs and 3D PPAFs electrode. The reason was that the deposition time was too short to cover the substrate electrode and too long to made the best of internal surfaces towards ethanol electrooxidation.
As we known, ethanol electrooxidation (EOR) is widely used since ethanol is less toxic than methanol and can be easily produced in great quantity by the fermentation of sugar containing raw materials. Furthermore, Hathoot and co-workers have found that Pd incorporated into the poly1, 2-diaminoanthra-quinone/glassy carbon (p1, 2-DAAQ/GC) electrode shows excellently higher activity than Pt for ethanol electrooxidation in alkaline media[31]. Consequently, the Pd and Pd-based catalyst used for ethanol electrooxi-dation in alkaline media have been received more and more attention. In this work, the as-prepared 3D porous mirco/nanostructures were also employed as electrocatalyst to study its electrochemical behavior of oxidizing ethanol.
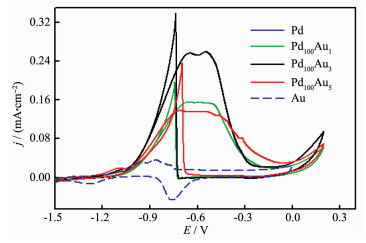
Here, to compare the catalytic activity of 3D PPF and PGPPs, the performances of Pd and 3D porous PdAu alloy electrocatalysts for ethanol oxidation in 0.5 mol·L-1 NaOH solution were tested. The ECAS of as-prepared electrodes were measured by CV in 0.5 mol·L-1 H2SO4 at 100 mV·s-1 (Fig. 5(a)) and current densities normalized by taking 424 μC·cm-2 as the charge density for the formation of a fully covered Pd oxide layer. As we known, the 3D porous Pd films are active for EOR. Blue line curve shows the first voltammogram run with a 3D PPF electrode in 0.5 mol·L-1 NaOH (Fig. 6 (dash line)) and containing 0.5 mol·L-1 ethanol at a scan rate of 20 mV·s-1. The comparison of two CVs clearly showed that EOR on Pd catalyst occured in the potential region which was from -1.2 to -0.08 V (vs SMSE), which partially overlaped with the 3D PPFs surface oxide formation. The reverse scan peak represented the removal of carbonaceous species not completely oxidized in the forward scan. It is worth noting that the whole pattern is reminiscent of that observed in the ethanol electrooxidation process on Pt-based electro-catalysts in acidic media[19-20]. However, all the alloyed PdAu catalysts are more favorable to ethanol oxidation reaction. The results showed synergistic effect of PdAu bimetallic surfaces because the electrooxidation onset potential was more negative compared with that on 3D PPF electrode which is shown in Fig. 6. The change in the onset potential showed an improvement in the kinetics. Furthermore, the sample of Pd100Au3 gave the best performance for EOR and the peak current density was 2.2 times higher for the reaction as compared to that on pure Pd catalyst. Preliminary results indicated that the catalytic activity of PdAu alloy catalysts can be significantly enhanced through adjusting the surface structures by changing the Au content in alloys. The magnitude of the forward anodic peak current densities of alloy samples cannot be due to the different surface areas because the current values were normalized with respect to the ECSAs, but rather the promotional effect of Au on Pd catalytic activity. The enhanced catalytic activity of the PdAu bimetallic porous nanostructures is consistent with the results of work already published in several studies about PdAu alloy[32] and the alloy system of Pd and Pt[25].
Figure 6
Figure 6.
CVs of 3D PPF in 0.5 mol·L-1 NaOH (dash line) and 3D PPF and PPAFs in 0.5 mol·L-1 NaOH+0.5 mol·L-1 CH3CH2OH with scan rate of 20 mV·s-1
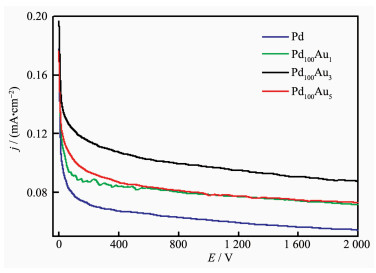
Long-term oxidation of ethanol was performed at a constant voltage of -0.8 V vs SMSE for 3D porous Pd and 3D porous PdAu electrodes, as shown in Fig. 7. The porous PdAu alloys demonstrated a relatively higher current density for a long time. Although the current densities of PdAu alloys dropped to approximately 50% of their initial value after 200 s, their remained steady at that level, showing much less decay than pure Pd electrodes. It was also found that the Pd100Au3 electrode exhibited the highest initial current and the slowest current decay, and the oxidation current was still considerably higher than those obtained at the others at a time of 2 000 s. These results were consistent with the CVs measurements mentioned above, further confirmed the relatively better tolerance to the intermediate species and higher catalytic activity of PdAu electrocatalysts toward EOR than that of pure Pd catalyst.
Figure 7
Figure 7.
Chronoamperometric curves for the 3D PPF and PPAFs in 0.5 mol·L-1 NaOH+0.5 mol·L-1 CH3CH2OH at -0.8 V vs SMSE
As for the origin of the enhanced EOR catalytic activity of the porous PdAu alloy catalysts, we assumed that the following factors play a key role. The first and most important, it is well known that the electrocatalytic activity of precious metal catalysts for fuel molecules electrooxidation is strongly dependent on the structures of catalysts, such as the component, particle size and morphology[33-34]. On the other hand, oxidation of ethanol involves multistep adsorption and electron transfer. The generally accepted oxidation sequence for ethanol oxidation in alkaline media reads, herein, CH3COads represents the adsorption of methanol, and OHads represents the adsorption of hydroxide ion.
Multistep adsorption requires large numbers of active sites on the electrode surface. The unique features of high surface area of 3D PPFs and PPAFs outermost surfaces for charge and mass (gas) transport meet this requirement well thus is favorable for the oxidation of ethanol. In addition, recent investigations indicated the structures of PdAu nanostructures highly depend on the preparation methods[11-12, 32]. Different preparation routes are result in different bimetallic PdAu surface morphologies and catalytic activities.
Second, it is well known that the physical properties of bimetallic nanomaterials depend strongly on their crystalline structures, for instance whether or not the two monometallic elements are chemically segregated or intimately alloyed. However, fundamental understanding of catalytic activity correlations in electronic property have been received much attention recently. Interestingly, an upshift of Au4f was observed for PdAu in the reference[15], probably because of the part of Au unalloyed. The lower BEs for both Pd3d3/2 and Au4f7/2 are consistent with the net charge flowing into Au (higher Au s or p electron densities) and Pd (gaining d electrons from Au), indicating the electronic structure of the surface Pd atoms to be modified via alloying with Au, and vice versa. It is generally agreed that the chemisorption of CO on Pd involves the donation of an electron pair from the σ* antibonding orbitals of CO to the unfilled 5d orbitals of Pd. A back donation of electrons from the Pd metal to the CO orbitals further stabilizes their interaction. Thus, the dative electron donation from CO to Pd is a prerequisite for strong CO chemisorption. Nevertheless, the d-charge transfer from Au to Pd in the PdAu alloy system led to a substantial increase in the electron density around the Pd sites. This increase would result in the weaker chemisorption of CO because of the lower d-band center compared with pure Pd. Briefly, there exists a stronger electron donation to the CO band by a Pd absorption site surrounded by Au atoms in the bimetallic alloy surface than that from the mono-metallic Pd surface which weaker chemisorption of CO. The electronic influence of Au on Pd was further confirmed by Wang and co-workers[35], and such enhancements of catalytic activity have previously been seen for many PdAu systems[11-12, 32, 35] and are thought to be due to synergistic electronic effects in which charge flow between Au and Pd[32, 35], thereby may result in an enhancement in activity. In conclusion, the enhanced catalytic acitivity of PdAu alloy originate from comprehensive effect which include structures and electronic properties of catalysts. On the basis of these analyses, it is not unexpected that the nanoporous PdAu alloy nanowires can exhibit distinctly enhanced catalytic performance toward methanol oxidation and a potential candidate for the application in direct ethanol fuel cells.
3.
Conclusions
In summary, a simple and fast hydrogen bubble dynamic template electrodeposition method is developed for the synthesis of the three-dimensional porous PdAu films. The introduction of Au into the Pd matrix can give rise to the added advantages of the synergetic and electronic effect resulting in improved reactivity of the bimetallic catalysts. The morphology, structure, composition, and electrocatalytic activity of the synthesized PdAu alloys can be easily controlled through varying the molar ratio between Pd and Au precursor. The sample of Pd100Au3 electrocatalyst with Pd/Au molar ratio of 97.85:2.15 in surface exhibited excellent electrocatalytic properties toward ethanol. The developed approach can be a useful method for preparing PdAu electrocatalysts which potentially can be used in the fields of catalysis, fuel cells, and other related fields.
[1]
Zheng L Z, Li J H. J. Phys. Chem. B, 2005, 109:1108-1112 doi: 10.1021/jp0456234
Figure 1
SEM images of as-prepared 3D PPF samples (a, c, e) and 3D porous Pd100Au3 films (b, d, f) by co-deposition along with H2 evolution at a constant potential of -4 V for 200 s taken with magnifications 200 (a, b), 3 000 (c, d) and 30 000 (e, f)
Figure 5
(a) CV curves of as-prepared 3D PPF and PPAFs electrodes (deposition time, 200 s; electrolyte, 0.5 mol·L-1 H2SO4 aqueous solution; sweep rate, 100 mV·s-1; 25 ℃); (b) Dependence of ECSAs on the electrodeposition time


 下载:
下载:

 下载:
下载:








