引用本文:
熊威, 冯建勇, 马为民, 赵劲, 李朝升, 邹志刚. 基于无机材料-微生物复合的半人工光合作用[J]. 无机化学学报,
2019, 35(9): 1521-1534.
doi:
10.11862/CJIC.2019.186

Citation: XIONG Wei, FENG Jian-Yong, MA Wei-Min, ZHAO Jing, LI Zhao-Sheng, ZOU Zhi-Gang. Semi-artificial Photosynthesis Based on Inorganic Material-Microbe Hybrids[J]. Chinese Journal of Inorganic Chemistry, 2019, 35(9): 1521-1534. doi: 10.11862/CJIC.2019.186
Citation: XIONG Wei, FENG Jian-Yong, MA Wei-Min, ZHAO Jing, LI Zhao-Sheng, ZOU Zhi-Gang. Semi-artificial Photosynthesis Based on Inorganic Material-Microbe Hybrids[J]. Chinese Journal of Inorganic Chemistry, 2019, 35(9): 1521-1534. doi: 10.11862/CJIC.2019.186
基于无机材料-微生物复合的半人工光合作用
摘要:
基于无机材料-微生物复合半人工光合系统是在自然光合作用和人工光合作用研究进展到一定阶段,为克服各自的缺陷,实现微生物与无机材料优势互补而发展出来的一种研究体系。该体系的主要优势是将微生物的催化选择性与无机材料的光响应性结合起来,旨在解决人工光合作用体系催化选择性差的问题。目前,可以通过光催化剂-微生物复合和电极-微生物复合来实现基于无机材料-微生物复合的半人工光合作用。本文围绕基于无机材料-微生物复合的半人工光合作用,依次从半人工水氧化、半人工光合还原和材料-微生物界面等方面做了系统的阐述,重点介绍基于电极-微生物复合的半人工光合体系研究进展,对基于无机材料-微生物复合的半人工光合作用的领域现状做了分析和总结,并且对该领域的前景进行了展望。
English
Semi-artificial Photosynthesis Based on Inorganic Material-Microbe Hybrids
Abstract:
Semi-artificial photosynthetic system based on inorganic material-microbe hybrid is a research system developed in the aspects of natural and artificial photosynthesis research to a certain stage, in order to overcome their respective defects and achieve complementary advantages of microbe and inorganic material. The main advantage of this system is the conjunction of catalytic selectivity of microbe and photo-response property of inorganic materials, which aim to solve the problem of poor catalytic selectivity of artificial photosynthetic system. At present, microbe-based semi-artificial photosynthesis can be achieved by photocatalyst-microbe hybrids and electrode-microbe hybrids. In this review, semi-artificial photosynthesis based on inorganic material-microbe hybrid is systematically expounded from semi-artificial water oxidation, semi-artificial photosynthetic reduction and material-microbe interface. The research progress of semi-artificial photosynthetic system based on electrode-microbe hybrids is emphatically introduced. The current situation of semi-artificial photosynthesis based on inorganic material-microbe hybrids has been analyzed and summarized, and the prospects of this field are outlooked.
-
Key words:
- inorganic material
- / artificial photosynthesis
- / microbe
- / photocatalysis
- / bio-catalysis
-
0. 引言
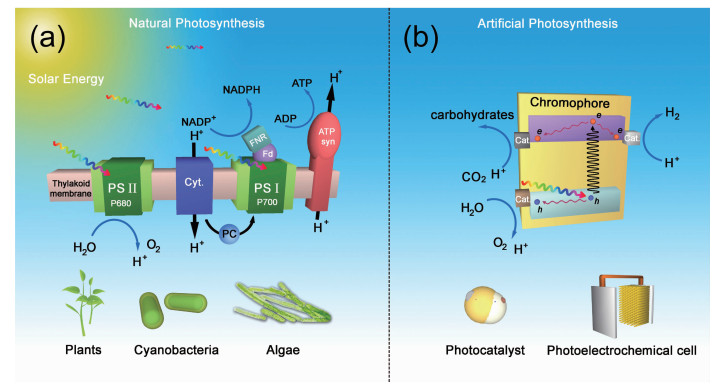
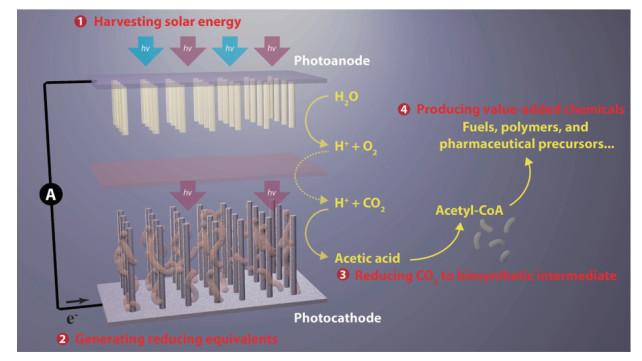
能源和环境危机已成为全球性的问题,而目前很多环境问题,恰是由于对化石能源的不合理利用造成的,因此应对能源与环境危机的关键在于如何解决能源问题。开发和利用清洁可再生能源作为应对能源和环境问题的重要解决方案已在全球范围内达成共识,而太阳能作为一种理想的可再生能源,实现太阳能到化学能的高效转换一直是科学家们追求的目标[1-8]。自然界可以通过光合作用储存太阳能,在这个过程中利用太阳光将水和二氧化碳转变为碳水化合物和氧气。利用人造材料模拟自然光合作用也能实现太阳能-化学能的转化,但是人工光合作用系统目前主要以氢气的形式将太阳能储存下来[2, 6, 8],尚不能通过二氧化碳固定实现完整的卡尔文循环[9]。相比于自然光合系统,人工光合系统具有更强的可设计性和更大的太阳光波长利用范围,但是自然光合系统具有更强的催化选择性。因此,近年来一些研究人员开始尝试通过人工光合系统的功能组分(光吸收层,光电极)与自然的催化体系协同复合来实现太阳能到化学能的转化,为实现自然光合作用与人工光合作用的耦合提供了一种新的策略,这种新的太阳能-化学能转化模式通常被称为半人工光合作用。
在详细介绍半人工光合作用前,首先需要介绍一下光合作用。光合作用按时间先后顺序主要分为3个物理化学过程:光能捕获,电荷产生与分离,催化反应。在自然光合作用中,光系统Ⅰ(PSⅠ)和光系统Ⅱ(PSⅡ)通过叶绿素捕获光能并将电子泵至激发态,在PSⅡ的锰钙氧化团簇上形成空穴发生水氧化反应[10]。而处于激发态的电子传递到光系统PSⅠ后,与PSⅠ端的空穴结合,PSⅠ上的激发态电子则参与后续的暗反应过程,这就是所谓的“Z型机制(Z-scheme)”[11]。在理想条件下,这些过程的电荷分离量子效率接近100%。然而自然光合作用的效率在约20%的标准太阳光强度下就会达到光饱和,更强的光照会导致生物光损伤[12-13],并且光损伤的修复也要消耗能量,同时降低太阳能-生物质的转化效率。自然光合作用的催化反应是在多个酶共同参与下的多种代谢途径的综合表现,可以利用二氧化碳、氮气、水等小分子选择性的合成大分子产物。但是自然光合作用的生物质转化效率一般不超过6%(农作物在1%~2%,大多数植物为0.1%)[14],因为自然界的光合生物优先进行光合生长而不是进行高能代谢产物的合成。最近兴起的合成生物学手段为提高生物质转化效率提供了一种合理有效的光合生物改造途径[15-17]。此外,仿生无机纳米材料也可用于改造光合微生物[18-23]。
在人工光合作用方面,光捕获过程始于半导体材料的光吸收。这些半导体材料不仅可以实现宽太阳光谱吸收,还可以设计成叠层配置以实现太阳光谱的分段互补吸收,而植物的叶绿素仅能选择性捕获太阳光谱中的部分光,并且太阳光的强度相对于催化中心是过饱和的[24]。此外,这些半导体材料通过掺杂和异质结等手段可以实现高效的电荷分离,以及导电层的电荷传输。相比于自然光合系统,人工光合系统构成更加简易,更易于以模块化方式进行替换和改进。实际上,利用H2O和CO2产生燃料的人工光合实验装置已经实现了远高于普通光合生物的太阳能转化效率(大于10%)[25-29]。然而,上述人工光合系统需要使用昂贵的高纯度半导体材料,而且这些半导体材料在电解质溶液中长时间工作容易被分解或腐蚀,也没有自修复机制来缓解该问题,因此这种人工光合系统尚不具备大规模使用的可行性。基于目前人工光合系统的快速进展以及未来的前景,研究人员仍在努力探索和开发高效且价格合理的催化剂,以选择性地生成各种复杂的碳基和氮基化合物。另一方面,通过稳定反应中间体[30-33]和控制界面反应物浓度[34-35]来模拟酶的工程策略已显现出一定的前景,但尚未实现高效率和高选择性的基于CO2的C-C偶联还原反应。此外,虽然人工光合体系已经可以模拟自然光合作用的Z-scheme,但是几乎不能复制自然光合作用中跨膜pH梯度的产生或维持以驱动三磷酸腺苷(ATP)的合成(图 1)[36]。
图 1
鉴于上述2种系统的局限性,半人工光合系统寻求整合自然光合系统(催化反应选择性高)和人工光合系统(光吸收效率高)各自的优势,旨在实现2个系统的优势互补。在此背景下产生了以无机材料-酶复合和无机材料-微生物复合的形式来构建的2类半人工光合系统,每种系统都具有其自身的优势和局限性。一方面,酶是人工催化剂设计的灵感来源;当酶与光电极或光吸收剂复合时,它们可以接近100%的选择性高速转换电荷,催化在动力学上难以实现的一系列简单产物合成[37]。然而,分离酶的大量纯化过程(导致低可扩展性)及其固有的不稳定性(特别是在离体环境中)使得它们在太阳能转换中缺乏可持续的商业应用价值。另一方面,无机材料-微生物复合系统可以利用无机合成材料作为光吸收剂,微生物细胞作为“催化剂”,引导光能或电能通过微生物的代谢途径产生多种人工光合系统或材料-酶复合系统无法合成的复杂产物。酶和微生物的本质区别在于酶是蛋白质大分子,而微生物是生命体。本文主要介绍基于无机材料-微生物复合的半人工光合作用体系。这类体系中的无机材料主要以光催化材料为主,微生物主要以蓝藻和少数具有特殊代谢功能的细菌为主,无机材料以纳米颗粒或者光电极的形式与微生物细胞结合,并通过材料-细胞的界面电子传递,调控微生物细胞的代谢产物合成。此外,借助合成生物学手段可以进一步实现材料-细胞复合光合系统产物生成的多样性与选择性。除了这些优点之外,微生物的自我复制特性赋予材料-细胞复合光合体系潜在的应用前景。虽然材料-细胞复合体系具有材料-酶复合体系不具备的优势[38],但是它们目前还是一个相对未知的系统。与材料-酶复合光合体系一样,材料-细胞复合光合体系的太阳光谱利用率(相比于非光合细胞)通常也很高,这主要是与半人工光合体系的光吸收由半导体组件处理有关。尽管有上述诸多优势,材料与细胞之间的界面电荷转移是阻碍无机材料-微生物复合光合体系商业化前景的最大挑战之一。此外,电荷在微生物内部的传输也可能成为特定的代谢途径合成的障碍[39]。
自然光合作用和人工光合作用的研究都已经开展多年,但是任何一种途径都不能实现理想的太阳能-化学能转换和存储。在过去的10年里,半人工光合作用已经发展成为一个新兴的、涉及多学科的研究方向。其中,基于无机材料-酶复合的半人工光合体系是最早发展起来的,国内外都有课题组开展相关工作;而基于无机材料-微生物复合的半人工光合体系的研究是最近几年才逐渐兴起,美国目前处于领先地位。虽然国外在基于微生物的半人工光合作用领域已有多篇相关综述报道,但是国内对于这个领域的研究尚处于萌芽阶段[40-43]。因此,本文主要针对基于微生物的半人工光合作用的最新进展,进行系统的归纳和总结,并分析主要的问题,展望未来这个领域的发展趋势,希望能为国内的研究人员提供一些启发和帮助。
1. 基于无机材料-微生物复合的半人工水氧化
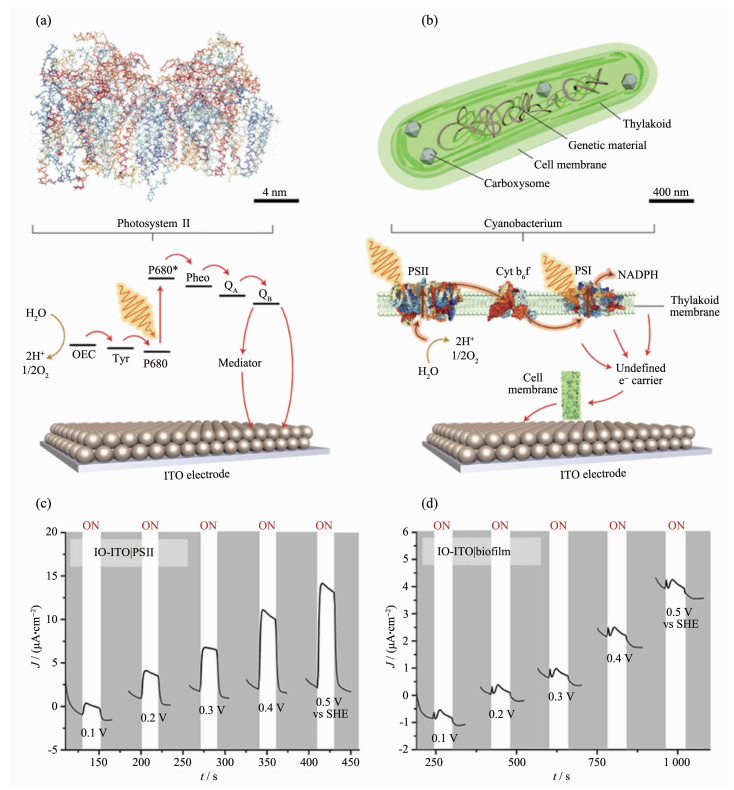
在光合作用过程中,水氧化生成氧气的反应是第一步化学反应。水是自然和人工光合作用的最终电子供体,并且水在自然界中大量存在,这使其成为生产太阳能燃料的最合适基质[44]。因此,理解水氧化反应的机制进而提高其效率对于实现高度功能化以及高效率的自然/人工光合作用是至关重要的。PSⅡ是自然界中唯一能够氧化水的酶,它同时也能有效地吸收光,分离光激发的电荷并将它们引导至所需的终点。实际上,这种酶负责所有大气中的氧气产生。该功能使PSⅡ成为探究水氧化反应机理的理想模型系统(图 2a),因为在人工光合系统中每一步反应的效率都是要追求极致的。但是PSⅡ对光非常敏感,每隔约15 min在体内修复一次,所以基于PSⅡ的电极也具有类似的运行寿命[45]。目前,除了与电极复合构成基于PSⅡ的半人工光阳极之外,PSⅡ也可以和光催化剂复合,实现半人工全水分解[46]。但是由于PSⅡ存在提取过程繁琐,离体稳定性差和光吸收限制等问题,基于PSⅡ的半人工光合作用系统更适合用来做模型研究,而不适合商业化应用[40]。
图 2
 图 2. (a) IO-ITO|PSⅡ半人工光阳极示意图[40]; (b) IO-ITO|蓝藻半人工光阳极示意图[40]; (c) IO-ITO|PSⅡ半人工光阳极在无外生介质和断续光照射下的多电位阶跃计时电流曲线[47]; (d) IO-ITO|蓝藻半人工光阳极在无外加导电介质和断续光照射下的多电位阶跃计时电流曲线[47]Figure 2. (a) Schematic of IO-ITO|PSⅡ semi-artificial photosynthetic electrode[40]; (b) Schematic of IO-ITO| cyanobacterium semi-artificial photosynthetic electrode[40]; (c) Stepped chronoamperometry scans of IO-ITO|PSⅡ in the absence of exogenous mediators (DET) and under chopped light irradiation[47]; (d) Stepped chronoamperometry scans of IO-ITO|cyanobacterium in the absence of exogenous mediators and under chopped light irradiation[47]
图 2. (a) IO-ITO|PSⅡ半人工光阳极示意图[40]; (b) IO-ITO|蓝藻半人工光阳极示意图[40]; (c) IO-ITO|PSⅡ半人工光阳极在无外生介质和断续光照射下的多电位阶跃计时电流曲线[47]; (d) IO-ITO|蓝藻半人工光阳极在无外加导电介质和断续光照射下的多电位阶跃计时电流曲线[47]Figure 2. (a) Schematic of IO-ITO|PSⅡ semi-artificial photosynthetic electrode[40]; (b) Schematic of IO-ITO| cyanobacterium semi-artificial photosynthetic electrode[40]; (c) Stepped chronoamperometry scans of IO-ITO|PSⅡ in the absence of exogenous mediators (DET) and under chopped light irradiation[47]; (d) Stepped chronoamperometry scans of IO-ITO|cyanobacterium in the absence of exogenous mediators and under chopped light irradiation[47]绝大多数用来做研究的PSⅡ都取自于蓝藻,而科学家们在研究中却发现一些蓝藻表面具有“纳米导线”可以将细胞内光合作用产生的电子传递到细胞表面。利用其电荷传输特性,将光合蓝藻贴在电极上培养繁殖可以实现自我维持的水氧化(图 2b)[47-48]。Erwin Reisner课题组将蓝藻连接到与其细胞尺寸匹配的大孔隙度(大于10 μm)反蛋白石结构氧化铟锡(IO-ITO)电极上,已经可以实现光阳极电流持续5d以上并且PSⅡ转换次数超过20 000(光照条件:1 mW·cm-2,λ=685 nm)[47]。有趣的是,该系统的性能随着时间的推移而增加,可能是蓝藻细胞与电极的连接状态越来越好的缘故。但是蓝藻基半人工光阳极的光电流密度(小于15 μA·cm-2)仍远低于PSⅡ基的半人工光合系统(图 2c,d)。在上述研究中,光态和暗态下的循环伏安测试对比还显示光诱导的氧化还原波可能与一种传递电荷的扩散介质有关,但目前学术界对此还存在争议。此外,Hasan等利用电极-PSⅡ复合体系研究的策略,将蓝藻和氧化还原聚合物同时固定在光电极表面,在44 mW·cm-2的光强照射下分别获得了8.6 μA·cm-2的直接光电流和48.2 μA·cm-2的介导光电流,但是可能会牺牲体系的寿命[49]。
虽然上述改进策略显示出一定的前景,并且蓝藻基电极的寿命(数周至数月)[50]超过PSⅡ基光阳极(数分钟)[45]和人工合成的光阳极(数小时至数天)[51],但是这种半人工光阳极的光电流输出仍须增加至少几个数量级才能在电力或太阳能燃料发电方面具有经济竞争力[48]。目前,基于无机材料-叶绿体复合的生物光电系统, 利用光阳极氧气生成和阴极氧气还原的催化环路,可以实现高达500 μA·cm-2的介导光电流[52]。因为蓝藻与叶绿体在生物进化上具有同源性,并且具有相似的结构和功能,所以使用蓝藻基半人工光阳极也有望实现500 μA·cm-2的光电流输出,但是该设计策略目前尚未在基于蓝藻的半人工光合系统中实现,这可能与用来构建该体系的蓝藻特性有关。蓝藻自身形成胞外电子的能力是构建该体系的基础。提高半人工光阳极的界面电荷转移率是将光电流密度提高到实用水平的关键。目前,已经在共聚焦荧光显微镜[53],电化学阻抗谱[54],表面增强拉曼/红外光谱[55-56]和纳米电极[57-58]等一些检测平台上建立起许多技术, 可以为半人工光阳极的界面电荷转移过程提供丰富的机理信息。例如,光谱检测扩散介质的分泌或膜结合细胞色素中的氧化还原变化, 可以揭示它们在细胞-电极的电荷转移机制中的作用[40]。蓝藻基半人工光阳极的另一个挑战是有效地引导通过细胞产生的电子仅流向水氧化途径,而不是流向生物质积累和代谢副反应的途径。通过仔细选择培养条件,或者使用特定的代谢化学抑制剂,甚至通过基因工程的手段,例如将蓝藻光合机构中的氨基酸替换,从而提高光化学量子产率和水氧化催化率[59],都有可能实现有效的电子传递路径调控。
基于无机材料-微生物复合的半人工水氧化反应,目前主要就是依靠蓝藻基半人工光阳极来实现,基本原理是蓝藻细胞内的PSⅡ发生水氧化反应时产生的电子流到细胞膜上,然后通过所谓的“纳米导线”传递到电极表面,最后在外电路中形成电流。相比于PSⅡ基半人工光阳极,蓝藻基半人工光阳极具有更高的稳定性、持久性以及更简便的制备过程,但是效率和光电流是它的劣势。就目前来看,基于电极-蓝藻复合的半人工水氧化离实际应用还很遥远。蓝藻细胞自身的光合作用效率,以及蓝藻细胞与电极界面的电荷转移效率是未来优化和改进蓝藻基半人工光阳极的关键。当然,其它一些化学反应动力学因素也是很重要的方面。其中,蓝藻细胞自身的光合作用效率可以通过合成生物学的手段和仿生无机纳米材料来改进[15, 18, 20],而蓝藻细胞与电极界面的电荷转移效率则可以通过对接触界面的调控来改善。此外,该体系可以为研究细胞向无机半导体材料表面的电荷转移提供支持。
2. 基于无机材料-微生物复合的半人工光合还原
自然光合作用包含光反应和暗反应2个阶段,在光反应阶段主要是水氧化和电子传递,而在暗反应阶段主要是以二氧化碳还原为核心的卡尔文循环。光反应为暗反应提供电子和能量来源,所以暗反应对于整个系统来说是还原反应。在半人工光合作用中,还原反应比水氧化反应具有更复杂的形式和更多变的产物,是整个体系的终端反应,也是半人工光合作用领域研究的焦点。目前,已经开发了基于无机材料-酶复合和无机材料-微生物复合的半人工光合还原系统,研究半人工光合还原系统的目标主要是理解和改进光合产氢、固碳和固氮的反应过程。基于无机材料-PSⅠ复合的半人工光合还原系统是最早开始研究的[60-62],目的是实现半人工光合产氢。随着氢酶提取和保存技术的进步,又出现了基于无机材料-氢化酶复合的半人工光合体系[63-68]。在此基础之上,将氢化酶换成二氧化碳还原酶或者固氮酶,也实现了半人工光合固碳和固氮[69-73]。但是基于无机材料-酶复合的半人工光合系统存在其固有的弊端,一是酶的提取和分离过程十分繁琐;二是离体后稳定性和持久性差等问题;三是材料与酶的复合可能影响酶的催化活性,而单一酶的催化就难以实现复杂产物的合成。因此,科学家们开始寻求具有相关还原反应代谢途径的微生物细胞与人工合成的无机半导体材料结合,构建基于无机材料-微生物复合的半人工光合还原系统。目前基于无机材料-微生物复合的半人工光合还原主要可以通过光催化剂-微生物复合体系和电极-微生物体系来实现。
2.1 光催化剂-微生物复合体系
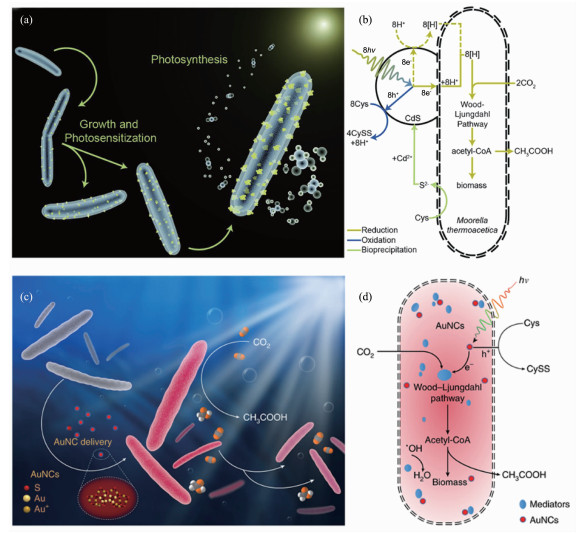
在最初的光催化剂-微生物复合的半人工光合体系研究中,杨培东课题组利用热醋穆尔氏菌(M. thermoacetica)的自我防御机制,将有生物毒性的镉离子(Cd2+)在细胞培养的过程中沉积到细菌细胞表面形成硫化镉(CdS)纳米颗粒(图 3a)[74]。在光照的条件下,来自CdS的光生电子穿过细胞膜通过Wood-Ljundahl途径成功地参与二氧化碳转化为乙酸的反应(图 3b),量子产率高达85%(光照条件:435~485 nm,5×1013 photon·cm-2 LED)[74]。受到先前量子点酶研究工作的启发,杨培东课题组采用泵浦探针瞬态吸收光谱对该系统的机理进行研究[75]。通过建立CO2光化学固定效率和瞬态吸收动力学的相关性与M. thermoacetica氢化酶表达量的函数关系,在不同时间尺度上证明直接的电荷转移途径依赖于显性的双氢化酶介导。为了消除对牺牲剂的依赖性,杨培东课题组又将M. thermoacetica-CdS杂化物与二氧化钛-锰-酞菁水氧化光催化剂在反应体系中共悬浮,也实现了CO2的固定,但是乙酸产率不高[76]。未来需要用无毒的光吸收剂替代CdS,通过选择透过性膜或区室化来防止氧气和活性氧物质伤害微生物,以及改善该系统在高太阳光强下的工作效率[45]。为了降低光催化材料与细菌细胞结合后产生的毒性,杨培东课题组在之前工作的基础上,将具有生物相容性的金纳米团簇引入M. thermoacetica细胞内部(图 3c)[77],依然起到CdS的光催化作用,并且在产生光生电子的同时具有抑制活性氧自由基产生的功能(图 3d),从而使得二氧化碳固定反应可以维持至少4 d。但是这个体系依然存在寿命问题,当细胞分裂形成新的细胞时,很难保证纳米金团簇还在细胞中。以上的研究说明,当微生物体系确定时,光催化剂的选择和设计对于整个半人工光合体系的性能有至关重要的影响。在选择和设计光催化剂时,一是要根据微生物细胞的生理特性选择生物毒性小的光催化剂,二是所选择的光催化剂能够以纳米材料的形式在细胞表面结合或者进入细胞内部。
图 3
 图 3. (a) 硫化镉-热醋穆尔氏菌复合体及其自光敏化太阳能转化[74]; (b)硫化镉-热醋穆尔氏菌复合体中太阳能-化学能转化的可能途径[74]; (c)金纳米团簇在热醋穆尔氏菌内部复合及其自光敏化太阳能转化[77]; (d)热醋穆尔氏菌-纳米金复合体中太阳能-化学能转化的可能途径[77]Figure 3. (a) Depiction of the CdS-M. thermoacetica hybrid system and self-photosensitization of it for solar-to-chemical conversion[74]; (b) Possible pathway for solar-to-chemical conversion of the CdS-M. thermoacetica hybrid system[74]; (c) M. thermoacetica photosensitized by intracellular gold nanoclusters for solar-to-chemical conversion[77]; (d) Possible pathway for solar-to-chemical conversion of the M. thermoacetica/AuNC hybrid system[77]
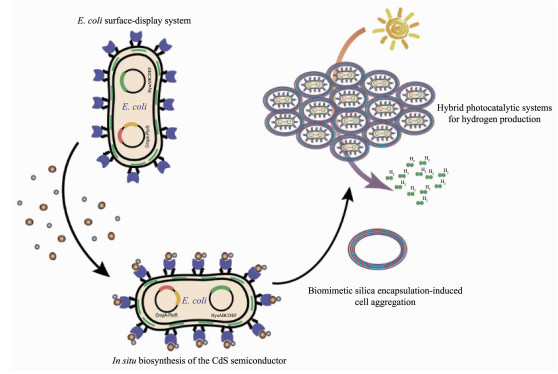
图 3. (a) 硫化镉-热醋穆尔氏菌复合体及其自光敏化太阳能转化[74]; (b)硫化镉-热醋穆尔氏菌复合体中太阳能-化学能转化的可能途径[74]; (c)金纳米团簇在热醋穆尔氏菌内部复合及其自光敏化太阳能转化[77]; (d)热醋穆尔氏菌-纳米金复合体中太阳能-化学能转化的可能途径[77]Figure 3. (a) Depiction of the CdS-M. thermoacetica hybrid system and self-photosensitization of it for solar-to-chemical conversion[74]; (b) Possible pathway for solar-to-chemical conversion of the CdS-M. thermoacetica hybrid system[74]; (c) M. thermoacetica photosensitized by intracellular gold nanoclusters for solar-to-chemical conversion[77]; (d) Possible pathway for solar-to-chemical conversion of the M. thermoacetica/AuNC hybrid system[77]通过以上的研究工作,可以总结出光催化剂-微生物复合的半人工光合作用的基本原理是光催化剂产生的光生电子被微生物利用,参与微生物的代谢反应,从而选择性合成某种代谢产物。因此,除了材料的选择和设计,微生物本身的性质对半人工光合体系的性能和效率的影响也是十分重要的方面,并且可以认为是决定性因素。最近兴起的合成生物学手段就是一种改造生物体的有效途径。赵劲课题组通过合成生物学手段构建了一种能在膜上表达重金属螯合蛋白PbrR和在细胞内部表达[NiFe]氢化酶的大肠杆菌,然后在大肠杆菌表面利用PbrR结合镉离子,形成CdS纳米颗粒,最后通过仿生硅矿包裹,实现了大肠杆菌的聚集,并诱导了大肠杆菌聚集体在有氧条件下的光合产氢(图 4)[78]。这项工作利用合成生物学方法与纳米技术,实现了对生物体的定向改造,为生物改造提供了新的思路。利用大肠杆菌的遗传操作特性,可以通过基因工程改造建立起一系列生物催化反应途径,但是使用无机光催化纳米材料与细菌结合,容易对细胞产生毒害作用,限制了催化反应效率。最近,Guo等使用简单的多酚化学将磷化铟纳米颗粒与转基因的酵母细胞(Saccharomyces cerevisiae Δzwf1)表面复合,以生成一种生物无机杂化体[79]。当白光照射在利用太阳能的酵母细胞上时,它们会捕获光生电子并将其用于再生化合物NADPH,这是一种在莽草酸合成过程中分解的辅助因子,因此它可以再次参与合成,从而促进莽草酸的生物合成[79]。该研究首次将光催化剂与微生物结合用于非能源物质的合成,拓展了半人工光合作用的应用领域,也为生物制造提供了新思路。
图 4

2.2 电极-微生物复合体系
与基于光催化剂-微生物复合的半人工光合还原体系相比,基于电极-微生物复合的半人工光合还原体系具有更好的可调性和更强的应用前景,并且对电极的修饰与改造比对微生物细胞表面的材料修饰更加具有操控性。该系统的最初发展得益于从微生物电合成化学池和半导体-电极界面的基础研究中获得的理解和经验[80]。杨培东课题组首先研究了希瓦氏菌(Shewanella oneidensis MR-1)在硅纳米线阵列上的识别[81],并且用氯化钠诱导了细菌在纳米线上的自组装[82]。随着基于材料-酶复合的半人工光合作用的研究深入,为了克服基于材料-酶的半人工光合体系的缺点,基于电极-微生物的半人工光合体系应运而生。这种系统的独特能力是通过微生物细胞内部代谢途径引导电极表面产生的还原物种(电子,氧化还原介质,氢气)转化为复杂的CO2和N2衍生产物[83]。根据还原物种的形式,这个体系可以分为集成式和分散式。所谓集成式,就是电极与微生物细胞是紧密结合的,还原物种的形式为电子,电极产生的电子直接传递给微生物参与CO2还原途径。而在分散式中,电极只是插入在微生物细胞的分散液中,并不和细胞界面连接,还原物种的形式为氢气或其他氧化还原介质,电极产生的电子先被用来产生氢气或者被氧化还原介质利用,然后再被微生物利用。因此,基于电极-微生物复合的半人工光合作用的基本原理可归纳为电极表面产生的还原物种进入微生物细胞,参与其内部代谢途径,转化为复杂的代谢产物。
对于集成式系统,实现电子在电极和细胞之间直接的传递需要两者之间具有良好的接触,因此高比表面积的电极对于提高电流密度就显得十分重要。杨培东课题组在国际上率先用硅纳米线阵列做光阴极与厌氧细菌Spormusa ovata (S. ovata)的细胞复合,二氧化钛纳米阵列作为光阳极,可以将CO2选择性地还原为CH3COOH(图 5)[84],并且法拉第效率达到90%。这种表面具有纳米线阵列的光阴极由于其独特的几何结构,将O2从其固定厌氧细菌的纳米线通道中排除,从而使整个系统可以在大气条件下工作,并在100 mW·cm-2的光照强度下,与TiO2光阳极配对时,以0.3 mA·cm-2的光电流密度驱动无辅助介质的CO2还原。微生物“催化剂”的自我复制性质使得操作寿命为200 h,并且通过基因工程改造使大肠杆菌将CH3COOH进一步升级为一系列更复杂的化学产物。
图 5

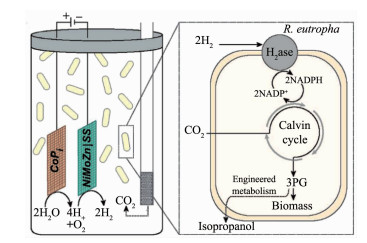
在分散式的体系中,Daniel G. Nocera课题组通过将硅基“人造叶”(阴极)[85]和InP-TiO2(阳极)[86]串联插入真氧产碱菌(Ralstonia eutropha)的细胞培养液中,在阴极产生氢气,通过细菌的氢气代谢过程还原CO2产生丁醇和甲烷(图 6)。但是这样的体系中,由于阳极会产生氧气,所以容易在体系中积累活性氧自由基,并且电极催化剂材料容易在反应的过程中腐蚀并释放出对微生物有毒害的金属离子。为了改进这个系统,Nocera课题组又开发了不会在溶液中产生显著活性氧物质或浸出金属离子的Co-P阴极-CoPi阳极催化体系[87],这是半人工系统向商业化应用迈出的有希望的一步。该电化学系统与Ge-GaAs-GaInP2三结光伏电池作为模型光伏电源和真氧产碱菌相结合,直接利用H2作为能量和还原物种输入,实现了持续5 d的太阳能驱动的CO2还原,并且太阳能-化学能转化效率可达到6%[87-88]。上述Co-P-CoPi系统进一步用于自养黄色杆菌(Xanthobacter autotrophicus)可以驱动N2向NH3的电化学固定[89]。这些自养黄色杆菌甚至可以作为肥料直接添加到土壤中,从而避免了产品提取和纯化的需要。鉴于固氮酶的氧敏感性,利用微生物作为保护性支架是使N2固定系统寿命延长的简便策略[90]。
图 6

虽然目前的研究进展是有应用前景的,但关键的限制是基于微生物的半人工光合系统的体积产品产率,而目前最优的系统能达到CH3COO-的产率为~1.3 g·L-1·d-1。为了使反应器和产物分离成本最低,扩大规模必需要提高产量。但是电化学池占用空间大,细胞与电极的低效连接以及单个物种持续生长的要求是基于微生物的半人工光合系统推向商业化应用的关键障碍。对微生物电合成等类似体系的研究已经表明电荷转移模式(直接、间接或两者兼有)是应变,环境,电极和时间依赖性的[91-93]。因此,首先需要深入理解界面化学和能量转移,然后通过代谢工程增加产品生成速率和化学多样性,最后将环境耐受性扩展到无污染的实验室之外的大规模培养条件。例如,如果半导体-电极界面处的能量转移通过H2发生,则可以通过使膜结合的氢化酶密度最大化来实现增加微生物H2吸收和产物产率[75]。最近,刘翀课题组发现使用生物相容性的全氟化碳纳米乳剂作为H2的载体,可以将S. ovata细菌的二氧化碳还原效率提高190%[94]。此外,电极极化或光照的变化如何引起基因表达和其他细胞行为的变化,以及如何有效地利用它对提高产率也是很重要的。同时降低催化反应过程中活性氧自由基和金属离子的积累,也是逐步走向大规模应用过程中需要克服和解决的问题。
3. 无机材料-微生物界面
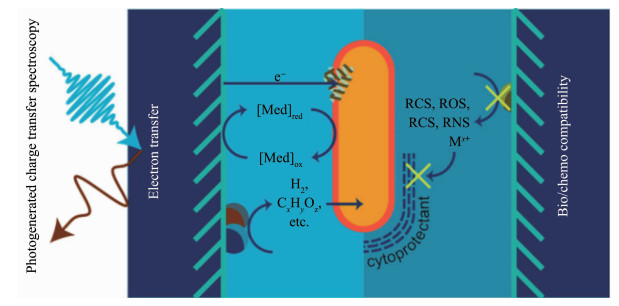
基于无机材料-微生物复合的半人工光合作用的基本原理是微生物光合作用产生的电子与人工合成的无机材料相互作用形成电流,或者无机材料在光照下产生的还原物种被微生物利用参与代谢反应。核心目标是无机材料与微生物催化体系的优势互补,材料与微生物的界面相互作用决定二者间的能量和电荷传递,因此深入理解材料-微生物的界面对于未来提高微生物基半人工光合系统的太阳能-化学能转化效率至关重要。在材料-微生物的界面相互作用中,有2个基础性问题:(1)材料与微生物之间的能量和电荷转移问题(图 7);(2)材料与微生物的相容性问题(图 7)[41]。
图 7

目前实现材料与微生物之间的电子转移主要有2种模式:直接转移和介导转移[41]。直接转移模式主要对应于光催化剂-微生物和集成式电极-微生物半人工光合体系,不需要外加氧化还原穿梭电对来传递电子,但是需要材料与微生物的紧密结合,这种模式对于材料-微生物的界面通常有特殊的要求,需要材料-微生物具有良好的匹配。而介导转移模式主要对应于分散式的电极-微生物半人工光合体系,利用可溶性的氧化还原穿梭电对将电极-溶液界面处产生的电子转化为还原物种提供给微生物[38, 87, 89]。这些还原物种继而将NAD(P)+还原为NAD(P)H,作为通用的生物电子供体,也可以产生ATP供生物催化反应使用。目前的介导转移系统可采用产氢电催化剂与H2氧化、CO2还原或N2还原细菌串联配对[87, 89, 91],利用H2作为还原物种,实现半人工光合还原。这些简易的系统具有高达~10%的太阳能-生物质能转化效率[87],比典型的植物太阳能-生物质能转化效率高一个数量级[95]。其它的氧化还原电对介质,例如甲酸盐[96-97],紫罗碱和吩嗪[98],也有一定的应用前景,但是它们对微生物的毒害性可能较大。单纯从电子转移效率来分析,直接转移模式比介导转移模式更有优势,因为氧化还原穿梭电对在传递电子的过程中不可避免存在能量损失,但是材料与微生物的直接接触对于彼此的相容性是很大的考验,细胞的生物活性以及无机光催化剂的活性都可能受到影响。此外,无机光催化剂的选择,电极的形貌结构和表面修饰都会对材料-微生物界面电子转移产生重要影响,这3个方面也是目前改进和优化系统的主要关注点。
考虑到无机材料与微生物直接接触可能发生的不利影响,如何在材料的生物相容性和微生物的化学相容性之间找到平衡点,一直以来都是这个领域的热点与难点。首先,材料的结构和形貌会影响其与微生物的结合进而影响微生物的活性。如将电极表面功能化或者纳米化会有利于微生物细胞在电极表面的吸附生长,而且二者接触面积的增大也会增加电流密度[99-100]。其次,任何一种含金属元素的催化剂在溶液中使用时,都不可避免的溶解产生金属离子,而绝大多数的金属离子对于微生物来说都是有毒性的。当微生物自身的活性下降后,内部的生物催化反应活性也可能受到影响,从而使整个系统的性能下降。但也有微生物对金属离子的响应是积极的,比如有些微生物本身就具备对某些重金属离子的抵抗作用[101-102],将此类金属元素以单质或者化合物的形式沉积在表面,能够促进材料-微生物的功能协同。此外,无机半导体材料在发生催化反应时,时常伴随着活性氧、活性氯和活性氮自由基[41],它们会对微生物细胞的活性产生毒害,因此需要在材料选择和设计时尽量避免此类情况的发生,并同时研究微生物对这些自由基活性种的响应机制。为了保护微生物,除了在无机半导体材料端做改进,通过化学和材料的手段对微生物细胞进行表面改进也是常用的方法,例如二氧化硅单细胞包裹微生物[18, 103],水凝胶包裹[104],金属有机框架材料包裹[105]等。
4. 研究总结与展望
半人工光合作用研究在近10年取得了迅猛的发展,初期的焦点主要集中在基于材料-酶复合的半人工光合作用,除了一些国外的课题组,国内的李灿院士团队在该领域也做出了有代表性的工作[5, 46, 106]。而基于无机材料-微生物复合的半人工光合系统研究则是在最近几年开始逐渐开展起来,目前处于领先地位的是美国的几个课题组,以Daniel G. Nocera和杨培东为代表。虽然他们已在这个领域做出了开拓性的工作,并且也在《科学》(Science),《自然》(Nature)子刊以及《美国国家科学院院刊》(PNAS)上发表多篇论文[40, 74, 77, 79, 85-87, 89],但是这个领域依然处于刚刚兴起的阶段[40-43]。最近,国内的麦立强课题组和美国的田博之课题组也开始关注这个领域中材料与生物的界面问题[42-43]。表 1列出了近5年来,基于无机材料-微生物复合的半人工光合作用的主要代表性工作。目前能够通过无机材料-微生物复合的半人工光合体系实现二氧化碳固定,生成乙酸、乙醇、莽草酸等小分子含碳有机物,但是效率、规模和成本尚未达到商业化应用的水平,因此还有很多科学和工程问题亟待解决。
表 1
 表 1 基于无机材料-微生物复合的半人工光合作用系统Table 1. Semi-artificial photosynthetic system based on inorganic-microbe hybrids
表 1 基于无机材料-微生物复合的半人工光合作用系统Table 1. Semi-artificial photosynthetic system based on inorganic-microbe hybrids 下载:
导出CSV
下载:
导出CSV
Microorganism Material System Hybrid Method Function Ref. Synechocystis sp.PCC 6803 ITO inverse opal electrodes Integrated combination Water oxidation, Biophotovoltaics [47] M. thermoacetica CdS nanoparticles Nanocoating CO2 reduction [74] M. thermoacetica CdS-TiO2-Mn-phthalocyanin Nanocoating CO2 reduction [75] M. thermoacetica Au nanoclusters Incorporation of nanoclusters CO2 reduction [77] Escherichia coli CdS/SiO2 Nanocoating and multicellular encapsulation H2 production [78] S. cerevisiae Δzwf1 InP nanoparticles Nanocoating CO2 reduction [79] S. aovata Silicon nanowire arrayed electrode Integrated combination CO2 reduction [84] R. eutropha NiMoZn-CoPi Distributed combination CO2 reduction [86] R. eutropha CoP-CoPi Distributed combination CO2 reduction [87] X. autotrophicus CoP-CoPi Distributed combination N2 fixation [89] S. aovata CoP-CoPi Distributed combination CO2 reduction [94] 微生物基的半人工光合系统中的一个核心问题就是材料-生物的界面相互作用,关键是能量与电荷在材料与微生物界面处的转移。材料与微生物的界面相互作用不仅影响微生物自身的性质,也会对材料性质产生影响。这种相互作用直接影响电荷在界面处的转移,因此提高界面电荷转移速率对提高微生物基半人工光合体系的太阳能-化学能转化效率至关重要。由于材料与微生物的界面相互作用主要取决于微生物的表面特性、合成材料的性质、微生物细胞与材料的结合形式,因此改进二者相互作用可以从3个方面入手:
(ⅰ)对微生物本身进行改造。在微生物改造方面,合成生物学具有越来越重要的作用[15-17]。虽然合成生物学一直致力于增强细胞的体内功能(例如合成回路和代谢途径的发展),但生物和非生物组分之间的界面是将来要解决的关键领域。为此,应用合成生物学的工具可以从头设计具有精细功能单元的多组分生物系统。该方法原则上可以使微生物适应从电极到工程代谢循环的高强度电荷通量。除了合成生物手段,通过仿生无机纳米材料对微生物进行功能化改造也是一种有效的途径[18-19, 22-23]。
(ⅱ)根据微生物的特性和最终的产物需求选择合适的材料体系。目前的半人工光合成体系,主要集中在合成小分子含碳有机物和氨等。如何进一步发挥生物体酶促反应的专一性、得到结构更复杂和具有更高价值的产物是未来探索的一个重要方向,这方面需要基于对相关微生物代谢途径和分子机制的理解,结合化学与材料的手段构建合适的半人工光合体系。
(ⅲ)改变材料-微生物的复合形式。未来的机遇还在于将合成材料与生物系统整合在一起,以产生能量转换和催化循环的新途径[42]。不同的复合形式会产生不同的材料-微生物相互作用,而不同的相互作用对微生物的代谢活动可能产生不同的影响。由于材料-微生物的界面是半人工光合体系的核心,因此材料-生物界面的原位表征技术是探索材料-微生物复合形式的重要基础。
总之,半人工光合作用是自然光合作用和人工光合作用交叉互补的一个体系,它的出现是自然光合作用和人工光合作用研究进展到一定阶段而产生的,未来的发展不仅取决于两个领域的研究水平和和认知程度,更取决于二者的融合程度。虽然基于无机材料-微生物复合的半人工光合作用目前尚处于概念验证阶段,但是过去5年的研究已经为基于无机材料-微生物的半人工光合作用未来的发展奠定了坚实的基础,未来这个领域在现有的基础上还有进一步完善和认识的空间。特别是微生物基的半人工光合体系可以作为模型体系来探索一些在纯自然光合作用或纯人工光合作用中无法用实验研究的问题,在新的微生物体系、材料体系与结构、材料-微生物结合方式等方面更加具有广阔的探索前景。此外,在一些特殊环境条件(例如海洋或太空航行)下,半人工光合系统可能存在更多的应用途径,但必须充分考虑资源管理和材料可回收性。在半人工光合体系迈向商业化应用的过程中,目标产物与生物催化体系的分离也是需要解决的问题。
-
-
[1]
Nocera D G. Acc. Chem. Res., 2017, 50(3):616-619 doi: 10.1021/acs.accounts.6b00615
-
[2]
Li Z S, Luo W J, Zhang M L, et al. Energy Environ. Sci., 2013, 6(2):347-370 doi: 10.1039/C2EE22618A
-
[3]
Li Z S, Feng J Y, Yan S, et al. Nano Today, 2015, 10(4):468-486 doi: 10.1016/j.nantod.2015.06.001
-
[4]
Tu W G, Zhou Y, Zou Z G. Adv. Mater., 2014, 26(27):4607-4626 doi: 10.1002/adma.201400087
-
[5]
Wang W Y, Wang H, Zhu Q J, et al. Angew. Chem. Int. Ed., 2016, 55(32):9229-9233 doi: 10.1002/anie.201604091
-
[6]
Yao T T, An X R, Han H X, et al. Adv. Energy Mater., 2018, 8(21):1800210 doi: 10.1002/aenm.201800210
-
[7]
Li X B, Tung C H, Wu L Z. Nat. Rev. Chem., 2018, 2(8):160-173 doi: 10.1038/s41570-018-0024-8
-
[8]
陈雅静, 李旭兵, 佟振合, 等.化学进展, 2019, 31(1):38-49 http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201901020.htmCHEN Ya-Jing, LI Xu-Bing, TONG Zhen-Ho, et al. Progress in Chemistry, 2019, 31(1):38-49 http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201901020.htm
-
[9]
Cook T R, Dogutan D K, Reece S Y, et al. Chem. Rev., 2010, 110(11):6474-6502 doi: 10.1021/cr100246c
-
[10]
Herek J L, Wohlleben W, Cogdell R J, et al. Nature, 2002, 417(6888):533-535 doi: 10.1038/417533a
-
[11]
Barber J. Chem. Soc. Rev., 2009, 38(1):185-196 doi: 10.1039/B802262N
-
[12]
Zhu X G, Long S P, Ort D R. Current Opin. Biotechnol., 2008, 19(2):153-159 doi: 10.1016/j.copbio.2008.02.004
-
[13]
Zhu X G, Long S P, Ort D R. Annu. Rev. Plant Biol., 2010, 61:235-261 doi: 10.1146/annurev-arplant-042809-112206
-
[14]
Blankenship R E, Tiede D M, Barber J, et al. Science, 2011, 332(6031):805-809 doi: 10.1126/science.1200165
-
[15]
Jagadevan S, Banerjee A, Banerjee C, et al. Biotechnol. Biofuels, 2018, 11(1):185 doi: 10.1186/s13068-018-1181-1
-
[16]
Kong F, Yamaoka Y, Ohama T, et al. Plant Cell Physiol., 2019, 60(6):1184-1196 doi: 10.1093/pcp/pcz022
-
[17]
Kung Y, Runguphan W, Keasling J D. ACS Synth. Biol., 2012, 1(11):498-513 doi: 10.1021/sb300074k
-
[18]
Xiong W, Yang Z, Zhai H L, et al. Chem. Commun., 2013, 49(68):7525-7527 doi: 10.1039/c3cc42766h
-
[19]
Xiong W, Zhao X H, Zhu G X, et al. Angew. Chem. Int. Ed., 2015, 54(41):11961-11965 doi: 10.1002/anie.201504634
-
[20]
Jiang N, Yang X Y, Deng Z, et al. Small, 2015, 11(17):2003-2010 doi: 10.1002/smll.201402381
-
[21]
Léonard A, Rooke J C, Meunier C F, et al. Energy Environ. Sci., 2010, 3(3):370-377 doi: 10.1039/b923859j
-
[22]
Ko E H, Yoon Y, Park J H, et al. Angew. Chem. Int. Ed., 2013, 52(47):12279-12282 doi: 10.1002/anie.201305081
-
[23]
熊威, 唐睿康, 马为民, 等.无机化学学报, 2019, 35(1):1-24 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20190101&journal_id=wjhxxbcnXIONG Wei, TANG Rui-Kang, MA Wei-Min, et al. Chinese J. Inorg. Chem., 2019, 35(1):1-24 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20190101&journal_id=wjhxxbcn
-
[24]
Cheng W H, Richter M H, May M M, et al. ACS Energy Lett., 2018, 3(8):1795-1800 doi: 10.1021/acsenergylett.8b00920
-
[25]
Khaselev O, Turner J A. Science, 1998, 280(5362):425-427 doi: 10.1126/science.280.5362.425
-
[26]
Zhou X H, Liu R, Sun K, et al. ACS Energy Lett., 2016, 1(4):764-770 doi: 10.1021/acsenergylett.6b00317
-
[27]
Verlage E, Hu S, Liu R, et al. Energy Environ. Sci., 2015, 8(11):3166-3172 doi: 10.1039/C5EE01786F
-
[28]
Ager J W, Shaner M R, Walczak K A, et al. Energy Environ. Sci., 2015, 8(10):2811-2824 doi: 10.1039/C5EE00457H
-
[29]
Jia J Y, Seitz L C, Benck J D, et al. Nat. Commun., 2016, 7:13237 doi: 10.1038/ncomms13237
-
[30]
Azcarate I, Costentin C, Robert M, et al. J. Am. Chem. Soc., 2016, 138(51):16639-16644 doi: 10.1021/jacs.6b07014
-
[31]
Rosen B A, Salehi-Khojin A, Thorson M R, et al. Science, 2011, 334(6056):643-644 doi: 10.1126/science.1209786
-
[32]
Gong M, Cao Z, Liu W, et al. ACS Cent. Sci., 2017, 3(9):1032-1040 doi: 10.1021/acscentsci.7b00316
-
[33]
Kim C, Jeon H S, Eom T, et al. J. Am. Chem. Soc., 2015, 137(43):13844-13850 doi: 10.1021/jacs.5b06568
-
[34]
Hall A S, Yoon Y, Wuttig A, et al. J. Am. Chem. Soc., 2015, 137(47):14834-14837 doi: 10.1021/jacs.5b08259
-
[35]
Ma S, Sadakiyo M, Luo R, et al. J. Power Sources, 2016, 301:219-228 doi: 10.1016/j.jpowsour.2015.09.124
-
[36]
Su J Z, Vayssieres L. ACS Energy Lett., 2016, 1(1):121-135 doi: 10.1021/acsenergylett.6b00059
-
[37]
Armstrong F A, Hirst J. Proc. Natl. Acad. Sci. USA, 2011, 108(34):14049-14054 doi: 10.1073/pnas.1103697108
-
[38]
Sakimoto K K, Kornienko N, Yang P D. Acc. Chem. Res., 2017, 50(3):476-481 doi: 10.1021/acs.accounts.6b00483
-
[39]
Kumar A, Hsu L H H, Kavanagh P, et al. Nat. Rev. Chem., 2017, 1(3):0024 doi: 10.1038/s41570-017-0024
-
[40]
Kornienko N, Zhang J Z, Sakimoto K K, et al. Nat. Nanotechnol., 2018, 13(10):890-899 doi: 10.1038/s41565-018-0251-7
-
[41]
Sakimoto K K, Kornienko N, Cestellos-Blanco S, et al. J. Am. Chem. Soc., 2018, 140(6):1978-1985 doi: 10.1021/jacs.7b11135
-
[42]
Xu L, Zhao Y L, Owusu K A, et al. Chem., 2018, 4(7):1538-1559 doi: 10.1016/j.chempr.2018.04.004
-
[43]
Lee Y V, Tian B Z. Nano Lett., 2019, 19(4):2189-2197 doi: 10.1021/acs.nanolett.9b00388
-
[44]
Lewis N S, Nocera D G. Proc. Natl. Acad. Sci. USA, 2006, 103(43):15729-15735 doi: 10.1073/pnas.0603395103
-
[45]
Kato M, Zhang J Z, Paul N, et al. Chem. Soc. Rev., 2014, 43(18):6485-6497 doi: 10.1039/C4CS00031E
-
[46]
Wang W Y, Chen J, Li C, et al. Nat. Commun., 2014, 5:4647 doi: 10.1038/ncomms5647
-
[47]
Zhang J Z, Bombelli P, Sokol K P, et al. J. Am. Chem. Soc., 2018, 140(1):6-9 doi: 10.1021/jacs.7b08563
-
[48]
McCormick A J, Bombelli P, Bradley R W, et al. Energy Environ. Sci., 2015, 8(4):1092-1109 doi: 10.1039/C4EE03875D
-
[49]
Hasan K, Yildiz H B, Sperling E, et al. Phys. Chem. Chem. Phys., 2014, 16(45):24676-24680 doi: 10.1039/C4CP04307C
-
[50]
Darus L, Ledezma P, Keller J, et al. Photosynth. Res., 2016, 127(3):347-354 doi: 10.1007/s11120-015-0193-y
-
[51]
Bae D, Seger B, Vesborg P C K, et al. Chem. Soc. Rev., 2017, 46(7):1933-1954 doi: 10.1039/C6CS00918B
-
[52]
Bhardwaj R, Pan R L, Gross E L. Nature, 1981, 289(5796):396-398 doi: 10.1038/289396a0
-
[53]
Pirbadian S, Barchinger S E, Leung K M, et al. Proc. Natl. Acad. Sci. USA, 2014, 111(35):12883-12888 doi: 10.1073/pnas.1410551111
-
[54]
He Z, Mansfeld F. Energy Environ. Sci., 2009, 2(2):215-219 doi: 10.1039/B814914C
-
[55]
Millo D, Harnisch F, Patil S A, et al. Angew. Chem. Int. Ed., 2011, 50(11):2625-2627 doi: 10.1002/anie.201006046
-
[56]
Busalmen J P, Esteve-Núñez A, Berná A, et al. Angew. Chem. Int. Ed., 2008, 47(26):4874-4877 doi: 10.1002/anie.200801310
-
[57]
Jiang X C, Hu J S, Fitzgerald L A, et al. Proc. Natl. Acad. Sci. USA, 2010, 107(39):16806-16810 doi: 10.1073/pnas.1011699107
-
[58]
Ding M, Shiu H Y, Li S L, et al. ACS Nano, 2016, 10(11):9919-9926 doi: 10.1021/acsnano.6b03655
-
[59]
Vinyard D J, Gimpel J, Ananyev G M, et al. J. Am. Chem. Soc., 2014, 136(10):4048-4055 doi: 10.1021/ja5002967
-
[60]
Millsaps J F, Bruce B D, Lee J W, et al. Photochem. Photobiol., 2001, 73(6):630-635 doi: 10.1562/0031-8655(2001)073<0630:NPPPOH>2.0.CO;2
-
[61]
Evans B R, O'Neill H M, Hutchens S A, et al. Nano Lett., 2004, 4(10):1815-1819 doi: 10.1021/nl0493388
-
[62]
Grimme R A, Lubner C E, Bryant D A, et al. J. Am. Chem. Soc., 2008, 130(20):6308-6309 doi: 10.1021/ja800923y
-
[63]
Tran P D, Artero V, Fontecave M. Energy Environ. Sci., 2010, 3(6):727-747 doi: 10.1039/b926749b
-
[64]
Brown K A, Wilker M B, Boehm M, et al. J. Am. Chem. Soc., 2012, 134(12):5627-5636 doi: 10.1021/ja2116348
-
[65]
Wilker M B, Utterback J K, Greene S, et al. J. Phys. Chem. C, 2017, 122(1):741-750
-
[66]
Brown K A, Dayal S, Ai X, et al. J. Am. Chem. Soc., 2010, 132(28):9672-9680 doi: 10.1021/ja101031r
-
[67]
Caputo C A, Gross M A, Lau V W, et al. Angew. Chem. Int. Ed., 2014, 53(43):11538-11542 doi: 10.1002/anie.201406811
-
[68]
Hutton G A M, Reuillard B, Martindale B C M, et al. J. Am. Chem. Soc., 2016, 138(51):16722-16730 doi: 10.1021/jacs.6b10146
-
[69]
Woolerton T W, Sheard S, Reisner E, et al. J. Am. Chem. Soc., 2010, 132(7):2132-2133 doi: 10.1021/ja910091z
-
[70]
Brown K A, Wilker M B, Boehm M, et al. ACS Catal., 2016, 6(4):2201-2204 doi: 10.1021/acscatal.5b02850
-
[71]
Brown K A, Harris D F, Wilker M B, et al. Science, 2016, 352(6284):448-450 doi: 10.1126/science.aaf2091
-
[72]
Hickey D P, Lim K, Cai R, et al. Chem. Sci., 2018, 9(23):5172-5177 doi: 10.1039/C8SC01638K
-
[73]
Milton R D, Abdellaoui S, Khadka N, et al. Energy Environ. Sci., 2016, 9(8):2550-2554 doi: 10.1039/C6EE01432A
-
[74]
Sakimoto K K, Wong A B, Yang P D. Science, 2016, 351(6268):74-77 doi: 10.1126/science.aad3317
-
[75]
Kornienko N, Sakimoto K K, Herlihy D M, et al. Proc. Natl. Acad. Sci. USA, 2016, 113(42):11750-11755 doi: 10.1073/pnas.1610554113
-
[76]
Sakimoto K K, Zhang S J, Yang P D. Nano Lett., 2016, 16(9):5883-5887 doi: 10.1021/acs.nanolett.6b02740
-
[77]
Zhang H, Liu H, Tian Z Q, et al. Nat. Nanotechnol., 2018, 13(10):900 doi: 10.1038/s41565-018-0267-z
-
[78]
Wei W, Sun P Q, Li Z, et al. Sci. Adv., 2018, 4(2):eaap9253 https://www.ncbi.nlm.nih.gov/pubmed/29492458
-
[79]
Guo J L, Suástegui M, Sakimoto K K, et al. Science, 2018, 362(6416):813-816 doi: 10.1126/science.aat9777
-
[80]
Rabaey K, Rozendal R A. Nat. Rev. Microbiol., 2010, 8(10):706-716 doi: 10.1038/nrmicro2422
-
[81]
Jeong H E, Kim I, Karam P, et al. Nano Lett., 2013, 13(6):2864-2869 doi: 10.1021/nl401205b
-
[82]
Sakimoto K K, Liu C, Lim J, et al. Nano Lett., 2014, 14(9):5471-5476 doi: 10.1021/nl502946j
-
[83]
Claassens N J, Sousa D Z, Santos V A P M, et al. Nat. Rev. Microbiol., 2016, 14(11):692-706 doi: 10.1038/nrmicro.2016.130
-
[84]
Liu C, Gallagher J J, Sakimoto K K, et al. Nano Lett., 2015, 15(5):3634-3639 doi: 10.1021/acs.nanolett.5b01254
-
[85]
Torella J P, Gagliardi C J, Chen J S, et al. Proc. Natl. Acad. Sci. USA, 2015, 112(8):2337-2342 doi: 10.1073/pnas.1424872112
-
[86]
Nichols E M, Gallagher J J, Liu C, et al. Proc. Natl. Acad. Sci. USA, 2015, 112(37):11461-11466 doi: 10.1073/pnas.1508075112
-
[87]
Liu C, Colón B C, Ziesack M, et al. Science, 2016, 352(6290):1210-1213 doi: 10.1126/science.aaf5039
-
[88]
Liu C, Colón B E, Silver P A, et al. J. Photochem. Photobiol. A:Chem., 2018, 358:411-415 doi: 10.1016/j.jphotochem.2017.10.001
-
[89]
Liu C, Sakimoto K K, Colón B C, et al. Proc. Natl. Acad. Sci. USA, 2017, 114(25):6450-6455 doi: 10.1073/pnas.1706371114
-
[90]
Milton R D, Cai R, Sahin S, et al. J. Am. Chem. Soc., 2017, 139(26):9044-9052 doi: 10.1021/jacs.7b04893
-
[91]
Marshall C W, Ross D E, Fichot E B, et al. Environ. Sci. Technol., 2013, 47(11):6023-6029 doi: 10.1021/es400341b
-
[92]
Siegert M, Yates M D, Call D F, et al. ACS Sustainable Chem. Eng., 2014, 2(4):910-917 doi: 10.1021/sc400520x
-
[93]
Zhang T, Nie H R, Bain T S, et al. Energy Environ. Sci., 2013, 6(1):217-224 doi: 10.1039/C2EE23350A
-
[94]
Rodrigues R M, Guan X, Iiguez J A, et al. Nat. Catal., 2019, 2:407-414 doi: 10.1038/s41929-019-0264-0
-
[95]
Ort D R, Merchant S S, Alric J, et al. Proc. Natl. Acad. Sci. USA, 2015, 112(28):8529-8536 doi: 10.1073/pnas.1424031112
-
[96]
Li H, Opgenorth P H, Wernick D G, et al. Science, 2012, 335(6076):1596-1596 doi: 10.1126/science.1217643
-
[97]
Yishai O, Lindner S N, de la Cruz J G, et al. Curr. Opin. Chem. Biol., 2016, 35:1-9 doi: 10.1016/j.cbpa.2016.07.005
-
[98]
Moscoviz R, Toledo-Alarcón J, Trably E, et al. Trends Biotechnol., 2016, 34(11):856-865 doi: 10.1016/j.tibtech.2016.04.009
-
[99]
Xie X, Hu L, Pasta M, et al. Nano Lett., 2010, 11(1):291-296 https://www.ncbi.nlm.nih.gov/pubmed/21158405
-
[100]
Mink J E, Rojas J P, Logan B E, et al. Nano Lett., 2012, 12(2):791-795 doi: 10.1021/nl203801h
-
[101]
Cunningham D P, Lundie L L. Appl. Environ. Microbiol., 1993, 59(1):7-14 https://www.ncbi.nlm.nih.gov/pubmed/8439169
-
[102]
Wang B, Zeng C, Chu K H, et al. Adv. Energy Mater., 2017, 7(20):1700611 doi: 10.1002/aenm.201700611
-
[103]
Yang S H, Lee K B, Kong B, et al. Angew. Chem. Int. Ed., 2009, 48(48):9160-9163 doi: 10.1002/anie.200903010
-
[104]
Lee K Y, Mooney D J. Prog. Polym. Sci., 2012, 37(1):106-126 doi: 10.1016/j.progpolymsci.2011.06.003
-
[105]
Liang K, Richardson J J, Cui J, et al. Adv. Mater., 2016, 28(36):7910-7914 doi: 10.1002/adma.201602335
-
[106]
Li Z, Wang W Y, Ding C M, et al. Energy Environ. Sci., 2017, 10(3):765-771 doi: 10.1039/C6EE03401B
-
[1]
-

图 2 (a) IO-ITO|PSⅡ半人工光阳极示意图[40]; (b) IO-ITO|蓝藻半人工光阳极示意图[40]; (c) IO-ITO|PSⅡ半人工光阳极在无外生介质和断续光照射下的多电位阶跃计时电流曲线[47]; (d) IO-ITO|蓝藻半人工光阳极在无外加导电介质和断续光照射下的多电位阶跃计时电流曲线[47]
Figure 2 (a) Schematic of IO-ITO|PSⅡ semi-artificial photosynthetic electrode[40]; (b) Schematic of IO-ITO| cyanobacterium semi-artificial photosynthetic electrode[40]; (c) Stepped chronoamperometry scans of IO-ITO|PSⅡ in the absence of exogenous mediators (DET) and under chopped light irradiation[47]; (d) Stepped chronoamperometry scans of IO-ITO|cyanobacterium in the absence of exogenous mediators and under chopped light irradiation[47]
图 3 (a) 硫化镉-热醋穆尔氏菌复合体及其自光敏化太阳能转化[74]; (b)硫化镉-热醋穆尔氏菌复合体中太阳能-化学能转化的可能途径[74]; (c)金纳米团簇在热醋穆尔氏菌内部复合及其自光敏化太阳能转化[77]; (d)热醋穆尔氏菌-纳米金复合体中太阳能-化学能转化的可能途径[77]
Figure 3 (a) Depiction of the CdS-M. thermoacetica hybrid system and self-photosensitization of it for solar-to-chemical conversion[74]; (b) Possible pathway for solar-to-chemical conversion of the CdS-M. thermoacetica hybrid system[74]; (c) M. thermoacetica photosensitized by intracellular gold nanoclusters for solar-to-chemical conversion[77]; (d) Possible pathway for solar-to-chemical conversion of the M. thermoacetica/AuNC hybrid system[77]
表 1 基于无机材料-微生物复合的半人工光合作用系统
Table 1. Semi-artificial photosynthetic system based on inorganic-microbe hybrids
Microorganism Material System Hybrid Method Function Ref. Synechocystis sp.PCC 6803 ITO inverse opal electrodes Integrated combination Water oxidation, Biophotovoltaics [47] M. thermoacetica CdS nanoparticles Nanocoating CO2 reduction [74] M. thermoacetica CdS-TiO2-Mn-phthalocyanin Nanocoating CO2 reduction [75] M. thermoacetica Au nanoclusters Incorporation of nanoclusters CO2 reduction [77] Escherichia coli CdS/SiO2 Nanocoating and multicellular encapsulation H2 production [78] S. cerevisiae Δzwf1 InP nanoparticles Nanocoating CO2 reduction [79] S. aovata Silicon nanowire arrayed electrode Integrated combination CO2 reduction [84] R. eutropha NiMoZn-CoPi Distributed combination CO2 reduction [86] R. eutropha CoP-CoPi Distributed combination CO2 reduction [87] X. autotrophicus CoP-CoPi Distributed combination N2 fixation [89] S. aovata CoP-CoPi Distributed combination CO2 reduction [94]  下载: 导出CSV
下载: 导出CSV
-








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 52
- 文章访问数: 2650
- HTML全文浏览量: 579
