Table 1.
Crystal data and structural refinement summary of the complexes 1~4
Citation:
TANG Long, YIN Si-Yu, WANG Ying-Lu, SHI De-Qian, HOU Xiang-Yang, WANG Xiao, WANG Ji-Jiang. Syntheses, Structures and Properties of Four Transition Metal Complexes with 2, 4-Oxybis(benzoic acid) and N-Donor Ligands[J]. Chinese Journal of Inorganic Chemistry,
2019, 35(10): 1728-1736.
doi:
10.11862/CJIC.2019.170

基于2, 4-氧基双(苯甲酸)和含氮配体的四种过渡金属配合物的合成、结构与性能
摘要:
采用水热法合成了4种过渡金属配合物{[Mn(2,4-Hoba)2(bipy)(H2O)2]·2H2O}n(1),[Mn(2,4-oba)(phen)]n(2),[Co(2, 4-oba)(bimyb)0.5]n(3)和[Ni(2,4-oba)(bimyb)0.5]n(4)(2,4-H2oba=2,4-氧基双(苯甲酸),bipy=4,4'-联吡啶,phen=1,10-菲咯啉,bimyb=1,4-双(咪唑-1-甲基)苯),并用X射线单晶衍射、热重分析、红外光谱和元素分析等手段对其进行了表征。单晶结构表明:配合物1是一维链状结构,通过O-H…O氢键扩展为二维超分子结构。配合物2是一维波浪链状结构,通过芳香环π-π堆积作用扩展为二维波状网络结构。配合物3和4是由配体桥联双核桨轮单元产生的二维层状结构。此外,对配合物2~4的磁性进行了研究,磁交换耦合常数分别为-0.79、-8.97和-11.42 cm-1。根据配合物2~4的晶体结构,利用DFT-BS方法对配合物2~4的磁耦合行为进行了研究。结果表明,计算的交换耦合常数J与实验数据吻合较好。
English
Syntheses, Structures and Properties of Four Transition Metal Complexes with 2, 4-Oxybis(benzoic acid) and N-Donor Ligands
Abstract:
Four transition metal coordination polymers {[Mn(2, 4-Hoba)2(bipy)(H2O)2]·2H2O}n (1), [Mn(2, 4-oba)(phen)]n (2), [Co(2, 4-oba)(bimyb)0.5]n (3) and[Ni(2, 4-oba)(bimyb)0.5]n (4) (2, 4-H2oba=2, 4-oxybis(benzoic acid), bipy=4, 4'-bipyridine, phen=1, 10-phenanthroline, bimyb=1, 4-bis(imidazole-1-ylmethyl) benzene) were synthesized by hydrothermal reactions and characterized by single-crystal X-ray diffraction, thermogravimetric analyses, IR spectroscopy and elemental analysis. Complex 1 is a 1D chain structure, which is extended to a 2D supramolecular architecture by O-H…O hydrogen bonding. Complex 2 shows a 1D wavy chain structure, which is expanded to a 2D wavelike network through aromatic π-π stacking interactions. Complexes 3 and 4 are 2D layer structures produced by ligands bridging binuclear paddle-wheel units. The magnetic properties of 2~4 have also been investigated, and the exchange conpling constants were -0.79, -8.97 and -11.42 cm-1, respectively. According to the crystal structures, the magnetic coupling behavior of complexes 2~4 were studied by using hybrid DFT-BS method, and the result reveals that the calculated exchange coupling constants (J) are in good agreement with the experimental data.
-
Key words:
- coordination polymers
- / crystal structure
- / magnetic behavior
- / density functional theory
-
0. Introduction
The rational design and synthesis of coordination polymers is currently of significant interest not merely due to the diverse network topology but mainly due to these extended systems playing a significant role in catalysis, chirality, luminescence, magnetism, nonlinear optics, adsorption, and separation[1-4]. In this context, the predesigned and assembly of novel crystalline material by structure-directing factors, such as central metal ions, organic ligands, metal-ligand ratio, solvents, temperature, pH value, and other factors[5-8], have been validated and summarized. Among them, the critical factor for the construction of coordination polymers is the rational choice of organic building block. In our strategy, multidentate O- or N-donor ligands have been employed in the construction of coordination polymers[9-11].
With regard to the flexible ether-oxygen dicarboxylate ligands, such as 2, 2′-oxybis(benzoic acid), 2, 4-oxybis(benzoic acid)(2, 4-H2oba) and 4, 4′-oxybis(benzoic acid) have been investigated[12-17]. Up to now, a number of N-containing ligands have been widely employed as the second ligands for meeting the requirement of coordination geometries of metal ions or tuning the fine structure. Compared with rigid bridging pyridyl ligands, 1, 4-bis(imidazole-1-ylmethyl)benzene (bimyb) possesses a flexible structure and an excellent coordination ability, and has the potential to construct coordination polymers. Inspired by those ideas, we successfully obtained four new coordination polymers, namely, {[Mn(2, 4-Hoba)2(bipy)(H2O)2]·2H2O}n (1), [Mn(2, 4-oba)(phen)]n (2), [Co(2, 4-oba)(bimyb)0.5]n (3) and [Ni(2, 4-oba)(bimyb)0.5]n (4) (bipy=4, 4′-bipyridine, phen=1, 10-phenanthroline). They are characterized by thermogravimetric analyses and X-ray crystallography. In addition, the magnetic properties of 2~4 are also studied.
1. Experimental
1.1 Materials and chemical analysis
The ligands 2, 4-H2oba, bimyb, bipy and phen were purchased from Jinan Henghua Sci. & Technol. Co. Ltd.; all other reagents and solvents employed were commercially available and used without further purification. Elemental analyses were performed with a Perkin-Elmer 2400 CHN Elemental analyzer. Infrared spectra on KBr pellets were recorded on a Nicolet 170SX FT-IR spectrophotometer in a range of 400~4 000 cm-1. TG analyses were conducted with a Nietzsch STA 449C micro analyzer under atmosphere at a heating rate of 5 ℃·min-1. X-ray powder diffraction (PXRD) patterns were recorded on a Shimadzu XRD-7000 diffractometer analyzer, the working voltage of PXRD is 40 kV, the current is 40 mA, the radiation source is Cu Kα (λ=0.154 18 nm), and the scanning range is 20°~80°. The magnetic susceptibilities were obtained on crystalline samples using a Quantum Design MPMS SQUID magnetometer.
1.2 Computational details
All calculations have been processed in Gaussian 03 package[18]. The magnetic isotropic shielding tensors were carried out with the hybrid DFT-BS method on the basis of B3LYP function[19]. The experimentally determined geometries for the complete structures of complexes 1~3 were used for the calculation of the magnetic exchange coupling constants[20]. Neither varia-tion of the geometrical parameters nor the geometry optimization was attempted in this calculation because a small variation in the geometry can have a big effect on the calculated magnetic interaction parameters.
1.3 Synthesis of {[Mn(2, 4-Hoba)2(bipy)(H2O)2]· 2H2O}n (1)
A mixture of 2, 4-H2oba (0.025 8 g, 0.1 mmol), bipy (0.015 6 g, 0.1 mmol), Mn(OAc)2·4H2O (0.061 3 g, 0.1 mmol), and H2O (10 mL) was stirred evenly and heated in a 23 mL Teflon-lined autoclave at 140 ℃ for 4 days, followed by slow cooling (5 ℃·h-1) to room temperature. The resulting mixture was washed with H2O, and yellow block crystals were collected and dried in air. Yield: 56% (based on Mn). Elemental analysis Calcd. for C38H34MnN2O14(%): C 57.22, H 4.30, N 3.51; Found(%): C 57.54, H 4.34, N 3.56. IR (KBr, cm-1): 3 480(s), 3 044(s), 2 368(vs), 1 625(s), 1 446(w), 1 397(vs), 1 134m, 796(s), 727(w), 681(vs), 562(vs).
1.4 Synthesis of [Mn(2, 4-oba)(phen)]n (2)
Complex 2 was prepared as for 1 by using phen (0.1 mmol, 0.019 8 g) instead of bipy. Yellow crystals of 2 were obtained (Yield: 58% based on Mn). Elemental analysis Calcd. for C26H16MnN2O5(%): C 63.55, H 3.28, N 5.70; Found(%): C 63.67, H 3.34, N 5.86. IR (KBr, cm-1): 3 108 (m), 1 607(vs), 1 563(s), 1 506(s), 1 429(s), 1 254(w), 1 087(m), 871(m), 776(m), 664(m).
1.5 Synthesis of [Co(2, 4-oba)(bimyb)0.5]n (3)
A mixture of Co(NO3)2·6H2O (0.1 mmol, 0.029 g), 2, 4-H2oba (0.1 mmol, 0.025 8 g), bimyb (0.1 mmol, 0.024 g) and water (10 mL) was stirred for 30 min in air. The mixture was transferred to a 23 mL Teflon reactor and kept at 140 ℃ for 5 days under autogenous pressure, and then cooled to room temperature at a rate of 5 ℃·h-1. Red crystals of 3 were obtained (Yield: 54% based on Co). Elemental analysis Calcd. for C21H15CoN2O5(%): C 58.08, H 3.48, N 6.45; Found(%): C 58.24, H 3.57, N 6.53. IR data (KBr, cm-1): 2 924 (w), 1 646 (vs), 1 615 (m), 1 443 (m), 1 418(s), 1 225(m), 1 137(m), 1 074(m), 876(m), 787(m), 664(w).
1.6 Synthesis of [Ni(2, 4-oba)(bimyb)0.5]n (4)
The procedure is similar to that of 3, except that Co(NO3)2·6H2O was replaced by Ni(NO3)2·6H2O (0.1 mmol, 0.029 g). Green crystals of 4 were obtained in 56% yield based on Ni. Elemental analysis Calcd. for C21H15NiN2O5(%): C 58.11, H 3.48, N 6.45; Found(%): C 58.26, H 3.54, N 6.56. IR data (KBr, cm-1): 2 927 (w), 1 645 (vs), 1 612 (m), 1 442 (m), 1 416(s), 1 225(m), 1 134(m), 1 078(m), 876(m), 786(m), 665(w).
1.7 X-ray crystallographic studies
Diffraction intensities for the four complexes were collected at 293 K on a Bruker SMART 1000 CCD diffractometer employing graphite-monochromated Mo Kα radiation (λ=0.071 073 nm). A semi-empirical absorption correction was applied using the SADABS program[21]. The structures were solved by direct methods and refined by full-matrix least-squares on F2 using the SHELXS 2014 and SHELXL 2014 programs, respectively[22-23]. Non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed in geometrically calculated positions. The crystallographic data for complexes 1~4 are listed in Table 1, and selected bond lengths and angles are listed in Table S1(Supporting information).
Table 1

 下载:
导出CSV
下载:
导出CSV
Complex 1 2 3 4 Empirical formula C38H34MnN2O14 C26H16MnN2O5 C21H15CoN2O5 C21H15NiN2O5 Formula weight 797.61 491.35 434.28 434.06 Crystal system Monoclinic Monoclinic Monoclinic Monoclinic Space group P2/c P21/n P21/n P21/n a/nm 1.220 93(17) 0.795 92(7) 1.176 6(3) 1.172 07(12) b/nm 1.156 24(16) 1.546 14(14) 1.033 1(3) 1.020 41(10) c/nm 1.557 72(16) 1.871 30(17) 1.555 5(4) 1.568 53(14) β/(°) 122.053(8) 99.949(2) 103.927(5) 103.921(2) V/nm3 1.863 8(4) 2.268 2(4) 1.835 2(9) 1.820 8(3) Dc/(g·cm-3) 1.421 1.439 1.572 1.583 Z 2 4 4 4 μ/mm-1 0.426 0.623 0.973 1.103 Reflection collected, unique 9 116, 3 286 (Rint=0.020 1) 11 259, 4 006 (Rint=0.052 2) 8 969, 3 231 (Rint=0.033 8) 11 153, 4 408 (Rint=0.035 5) Data, restraint, parameter 3 286, 3, 258 4 006, 0, 307 3 231, 0, 262 4 408, 0, 262 Goodness-of-fit (GOF) on F2 1.050 1.030 1.015 1.001 Final R indices [I>2σ(I)] R1=0.036 3, wR2=0.104 2 R1=0.043 3, wR2=0.069 9 R1=0.035 4, wR2=0.086 0 R1=0.039 8, wR2=0.080 8 Largest difference in peak and hole/(e·nm-3) 474 and -209 199 and -200 354 and -264 428 and -256 CCDC: 1910923, 1; 1910924, 2; 1910926, 3; 1910927, 4.
2. Results and discussion
2.1 Description of the structure
2.1.1 Crystal structure of {[Mn(2, 4-Hoba)2(bipy)(H2O)2]·2H2O}n (1)
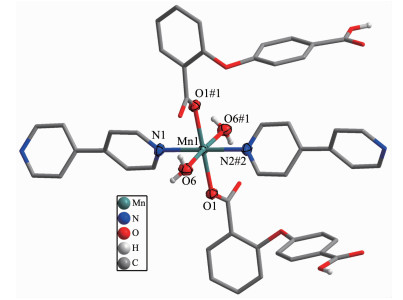
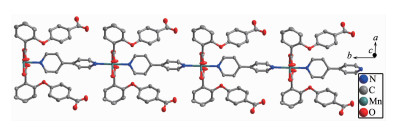
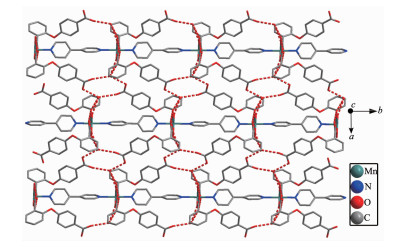
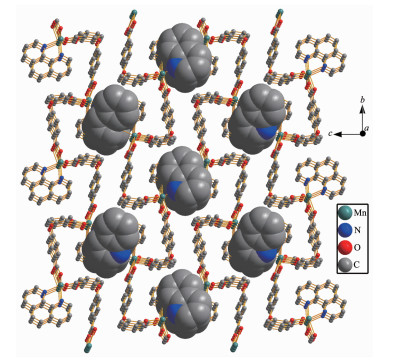
Single crystal X-ray diffraction analysis suggests that complex 1 consists of one Mn(Ⅱ) ion, two 2, 4-Hoba anions, one bipy molecule, two coordinated water and two free water molecules. Each Mn(Ⅱ) center is six-coordinated by two pyridyl nitrogen donors from two different bipy ligands and four oxygen atoms coming from two different Hoba ligands and two coordinated water molecules (Mn-N/O 0.217 99(13)~0.225 0(2) nm), forming a distorted MnN2O4 octahedral geometry (Fig. 1). The O/N-Mn-O/N bond angles are in a range of 87.36(4)°~180.0°. The adjacent Mn(Ⅱ) ions are bridged by bipy ligands to form an infinite 1D polymeric chain running along b-axis (Fig. 2). The 2, 4-Hoba ligands adopt a μ-unidentate (OCOO-) coordination mode (mode A in Scheme S1), two phenyl rings are severely bent, with the dihedral angle being 79.96°, and all 2, 4-Hoba ligands bristle out from two sides of the 1D chain. There are several kinds of hydrogen bonding in the structure: (a) hydrogen bonds between the coordinated water and free water molecule with O6…O7 distance of 0.301 1(2) nm; (b) hydrogen bonds between the coordinated water and carboxylate O atoms of the 2, 4-Hoba anion with the O6…O2 distance of 0.265 4(2) nm; (c) hydrogen bonds between the free water molecule and carboxylate O atoms of two 2, 4-Hoba anions with the O7…O1 and O7…O5 distance of 0.273 5(2) and 0.292 9(3) nm, respectively; (d) hydrogen bonds between carboxyl group of the 2, 4-Hoba anion and free water molecule with the O4…O7 distance of 0.266 5(3) nm (Fig.S1 and Table S2). Due to these strong intermole-cular O-H…O hydrogen bonds, the adjacent 1D chains are further extended to produce a 2D supra-molecular framework (Fig. 3).
Figure 1
 Figure 1. Coordination environment of Mn(Ⅱ) ion of complex 1
Figure 1. Coordination environment of Mn(Ⅱ) ion of complex 1Hydrogen atoms and the free H2O molecules are omitted for clarity except the coordination water; 40% ellipsoid probability level; Symmetry codes: #1:-x+1, y, -z+3/2; #2: x, y-1, z
Figure 2
Figure 3
2.1.2 Crystal structure of [Mn(2, 4-oba)(phen)]n (2)
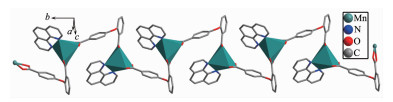
Single-crystal X-ray analysis reveals that complex 2 shows an infinite 1D wavy chain structure. Each Mn(Ⅱ) ion is coordinated to four oxygen atoms of two 2, 4-oba ligands and two nitrogen atoms of one phen ligand, forming a distorted octahedral geometry, as shown in Fig. 4. The bond lengths of Mn-N are 0.221 3(2) and 0.224 5(2) nm, the Mn-O bond lengths are in a range of 0.218 13(19)~0.223 57(19) nm, the O/N-Mn-O/N bond angles are in a range of 58.96(7)°~153.55(9)°. As compared to complex 1, the 2, 4-oba ligand of 2 adopts a bis(chelating bidentate) coordina-tion mode (Mode B in Scheme S1). The adjacent Mn(Ⅱ) centers are linked by 2, 4-oba ligands bridges to generate a 1D wavy chain with a Mn…Mn separation being 0.871 2 nm (Fig. 5). All phen ligands bristle out from two sides of the chain. Further through aromatic π-π stacking interactions between two phen ligands (centroid-to-centroid distance: 0.347 8 and 0.352 6 nm) (Fig.S2), the adjacent chains expand to a 2D wavelike network, as shown in Fig. 6.
Figure 4
 Figure 4. Coordination environment of Mn(Ⅱ) ion in complex 2
Figure 4. Coordination environment of Mn(Ⅱ) ion in complex 2All hydrogen atoms are omitted for clarity; Symmetry codes: #1: -x+3/2, y-1/2, -z+3/2
Figure 5
Figure 6
2.1.3 Crystal structures of [Co(2, 4-oba)(bimb)0.5]n (3) and [Ni(2, 4-oba)(bimb)0.5]n (4)
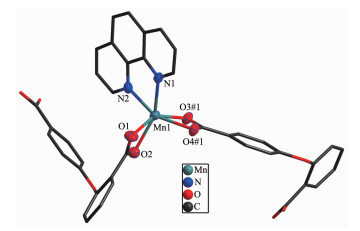
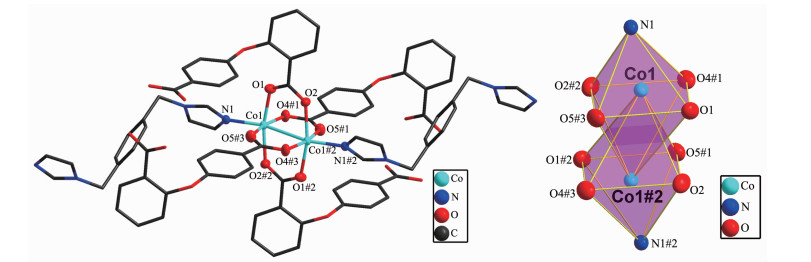
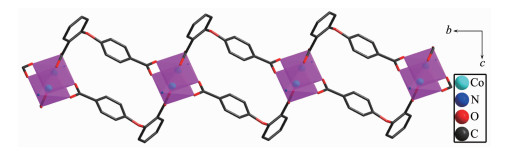
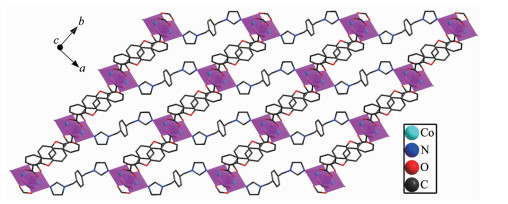
Single-crystal X-ray analysis reveals that complexes 3 and 4 are isomorphic, and the structure of complex 3 is described here. The asymmetric unit of 3 contains one Co(Ⅱ) ion, one 2, 4-oba dianion and half of the bimyb ligand with the metal-based building unit. In this building unit, each end of the 2, 4-oba ligand bridges a pair of Co(Ⅱ) ions to result in a binuclear Co(Ⅱ)-tetracarboxylate paddle-wheel of the type [Co2(OOCR)4][24], which comprises two cobalt ions and four di(monodentate) bridging carboxylates (Fig. 7). The bond lengths of Co-O/N are in a range of 0.201 2(2)~0.208 8(2) nm and the distances of Co1-Co1#2 is 0.275 35(9) nm. Each Co(Ⅱ) center has a pseudo-octahedral geometry. In complex 3, the coordination mode of 2, 4-oba ligand is different from those of complexes 1 and 2, since each 2, 4-oba ligand adopts a bis(bidentate bridging) coordination mode (Mode C in Scheme S1). The neighboring paddle-wheel units are linked by 2, 4-oba ligands bridges to generate a 1D loop shaped chain running along the b-axis (Fig. 8). Furthermore, through bimyb ligand bridging, the adjacent 1D chains are connected to produce a 2D wavy layer structure (Fig. 9). The structure of complex 4 is described in detail in the supporting information (Fig.S3~S5).
Figure 7
 Figure 7. Binuclear Co(Ⅱ)-tetracarboxylate paddle-wheel of 3
Figure 7. Binuclear Co(Ⅱ)-tetracarboxylate paddle-wheel of 3Hydrogen atoms are omitted for clarity; Symmetry codes: #1: x, y-1, z; #2:-x, -y+1, -z
Figure 8
Figure 9
2.2 PXRD and thermogravimetric analysis
X-ray powder diffraction (PXRD) was used to confirm the phase purity of the bulk material of 1~4 at room temperature (Fig.S6~S9). The experimental diffraction feature peaks of the bulk samples are fully consistent with the simulated patterns, indicating the good purity of 1~4. To study the thermal stability of 1~4, thermogravimetric (TG) analyses were performed on polycrystalline samples under a nitrogen atmosphere with a heating rate of 10 ℃·min-1 (Fig.S10~S13). The TG curve of 1 showed two steps of weight loss. The first weight loss in a range of 110~180 ℃ (Obsd. 9.1%, Calcd. 9.03%) is attributed to the loss of free water and coordinated water. The second weight loss of 83.9% in a range of 230~550 ℃ corresponds to the release of the 2, 4-oba and bipy ligands (Calcd. 84.08%). The TG curve of 2 suggested that no weight loss were observed until 220 ℃, above which significant weight loss(Obsd 88.6%) occurred and ended at about 430 ℃, indicating complete decom-position of the 2, 4-oba and phen ligands(Calcd. 88.82%). TG curve of 3 showed one step of weight loss, and the larger weight loss(Obsd. 86.6%) occurred in a range of 320~720 ℃, corresponding to the decomposition of the 2, 4-oba and bimyb ligands (Calcd. 86.43%). TG curve of 4 revealed only one large weight loss(Obsd. 86.6%) occurred in a range of 320~700 ℃, corresponding to the decomposition of the 2, 4-oba and bimyb ligands (Calcd. 86.48%).
2.3 Magnetic property
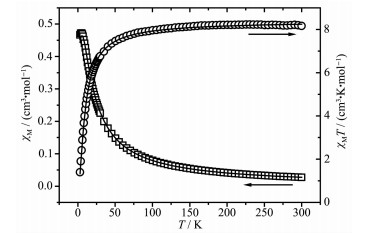
The magnetic properties of 2~4 were investigated in the 2~300 K temperature range at 1 000 Oe. The magnetic susceptibility data measured for 2 is shown in Fig. 10. As observed, the experimental χMT value at 300 K was 8.81 cm3·K·mol-1, which is close to that of two isolated spin-only Mn(Ⅱ) ions (8.75 cm3·K·mol-1, S=5/2). The χMT value of 2 remained almost constant from 300 to 120 K. As the temperature was lowered to 2 K, the value of χMT decreased down to a minimum value of 1.51 cm3·K·mol-1, which demonstrates intra-molecular antiferromagnetic interactions among the Mn(Ⅱ) ions. According to the crystal structure, the magnetic analysis of 2 was carried out using the theoretical expression of the magnetic susceptibility deduced from the spin Hamiltonian:
$ \hat H = - 2J{\hat S_{{\rm{Mn1}}}}{\hat S_{{\rm{Mn2}}}} $ [25-27], and the expression of the magnetic susceptibility for a binuclear unit is given by:$ {\chi _{\rm{M}}} = \frac{{2N{g^2}{\beta ^2}}}{{kT}}\frac{{{{\rm{e}}^{2J/\left( {kT} \right)}} + 5{{\rm{e}}^{6J/\left( {kT} \right)}} + 14{{\rm{e}}^{12J/\left( {kT} \right)}} + 30{{\rm{e}}^{20J/\left( {kT} \right)}} + 55{{\rm{e}}^{30J/\left( {kT} \right)}}}}{{1 + 3{{\rm{e}}^{2J/\left( {kT} \right)}} + 5{{\rm{e}}^{6J/\left( {kT} \right)}} + 7{{\rm{e}}^{12J/\left( {kT} \right)}} + 9{{\rm{e}}^{20J/\left( {kT} \right)}} + 11{{\rm{e}}^{30J/\left( {kT} \right)}}}} $
(1) Figure 10
 Figure 10. Thermal variation of χM and χMT for 2
Figure 10. Thermal variation of χM and χMT for 2Open points are the experimental data, and the solid line represents the best fit obtained from the Hamiltonian given in the text
J is the exchange coupling constant between adjacent Mn(Ⅱ) ions. The least-squares fit to the experimental data was found with J=-0.79 cm-1, g=2.01, and the agreement factor R, defined as R=∑[(χM)obs-(χM)calc]2/∑[(χM)obs]2, was 7.43×10-4. The small negative value of J further indicates that weak antiferromagnetic interaction exists among the Mn(Ⅱ) centers.
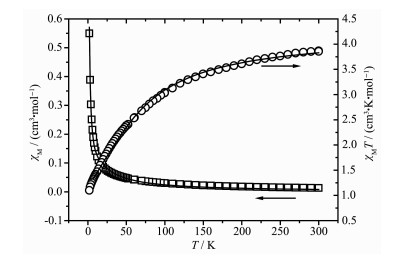
The magnetic susceptibility data measured for 3 is shown in Fig. 11. The experimental χMT value at 300 K was 3.86 cm3·K·mol-1, which is larger than the expected value (3.75 cm3·K·mol-1) of two isolated spin-only Co(Ⅱ) ions (S=3/2). As the temperature decreased, the value of χMT gradually decreased down to a minimum value of 1.09 cm3·K·mol-1 at 2 K, which suggests that antiferromagnetic interactions are operative. To simulate the experimental magnetic behavior, the magnetic susceptibility of 3 was fitted by the following expression deduced from the spin Hamiltonian
$ \hat H = - 2J{\hat S_{{\rm{Co1}}}}{\hat S_{{\rm{Co2}}}} $ , and the expression of the magnetic susceptibility for a dinuclear models is given by:${\chi _{\rm{M}}} = \frac{{2N{g^2}{\beta ^2}}}{{kT}}\frac{{{{\rm{e}}^{2J/\left( {kT} \right)}} + 5{{\rm{e}}^{6J/\left( {kT} \right)}} + 14{{\rm{e}}^{12J/\left( {kT} \right)}}}}{{1 + 3{{\rm{e}}^{2J/\left( {kT} \right)}} + 5{{\rm{e}}^{6J/\left( {kT} \right)}} + 7{{\rm{e}}^{12J/\left( {kT} \right)}}}} $
(2) Figure 11
 Figure 11. Thermal variation of χM and χMT for 3
Figure 11. Thermal variation of χM and χMT for 3Open points are the experimental data, and the solid line represents the best fit obtained from the Hamiltonian given in the text
The least-squares fit to the experimental data was found with J=-8.97 cm-1, g=2.14, and the agreement factor R was 2.38×10-4. The negative value of J further indicates antiferromagnetic interaction exists among the Co(Ⅱ) centers. According to the structure of 3, it could be presumed that the main magnetic interactions are between the paddle-wheel unit metal center, while the super-exchange interactions between Co(Ⅱ) ions through the bimyb bridge can be ignored due to the length of the bimyb ligands. The results indicate the there are typical antiferromagnetic and spin-orbit coupling interactions in the complex.
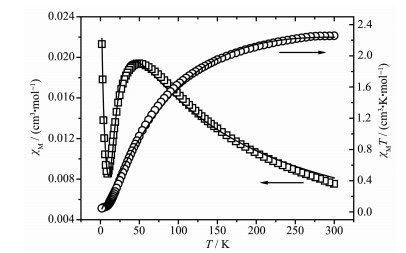
The magnetic susceptibility data measured for 4 is shown in Fig. 12. The experimental χMT value at 300 K was 2.26 cm3·K·mol-1, which is slightly higher than the expected value (2.0 cm3·K·mol-1, S=1) of two isolated spin-only Ni(Ⅱ) ions. As the temperature decreased, the χMT value gradually decreased till 2 K to reach a minimum value of 0.046 cm3·K·mol-1, manifesting a significant antiferromagnetic exchange between the magnetic Ni(Ⅱ) centers. The χM value first increased slowly to a broad maximum value of 0.0196 cm3·K·mol-1 at 49 K and then decreased rapidly to 0.008 8 cm3·K·mol-1 upon cooling to 8 K and finally rises rapidly until 2 K, indicating antiferromagnetic interaction between Ni(Ⅱ) ions (Ni-Ni 0.264 09(6) nm). The increase of χM below 9 K may be attributed to the paramagnetic impurity. The magnetic analysis was carried out using the theoretical expression for the magnetic susceptibility deduced from the spin Hamil-tonian
$ \hat H = - 2J{\hat S_{{\rm{Ni1}}}}{\hat S_{{\rm{Ni2}}}} $ , the expression for the magnetic susceptibility is given by:${\chi _{\rm{M}}} = \frac{{2N{g^2}{\beta ^2}}}{{kT}}\frac{{{{\rm{e}}^{2J/\left( {kT} \right)}} + 5{{\rm{e}}^{6J/\left( {kT} \right)}}}}{{1 + 3{{\rm{e}}^{2J/\left( {kT} \right)}} + 5{{\rm{e}}^{6J/\left( {kT} \right)}}}} $
(3) Figure 12
 Figure 12. Thermal variation of χM and χMT for 4
Figure 12. Thermal variation of χM and χMT for 4Open points are the experimental data, and the solid line represents the best fit obtained from the Hamiltonian given in the text
The best-fit parameters for the experimental data gave J=-11.42 cm-1, g=2.24, R=6.35×10-4. The large negative J also confirms antiferromagnetic interactions between two Ni(Ⅱ) ions.
2.4 Magnetic properties of DFT calculations
The DFT calculations have been widely proved to be one of the most efficient tools to investigate magnetic structure of transition metal complexes[28-30]. We used a phenomenological Heisenberg Hamiltonian
$\hat H = - J{\hat S_1}{\hat S_2} $ to describe the exchange coupling in a dinuclear complex, where the coupling constant J can be related to the energy difference between the lowest and highest spin states. For the case in which S1=S2, the coupling constant may be obtained by using the following equation (4)[31].$ {E_{{\rm{HS}}}}{\rm{ - }}{E_{{\rm{LS}}}} = - 2J{S_i}\left( {{S_i} + 1/2} \right) $
(4) Where EHS is the energy that corresponds to the state with the highest total spin; ELS corresponds to the state with the lowest total spin (S=0), and Si is the total spin on each metal atom.
When using DFT-based wave functions, a reasonable estimate of the energy corresponding to the low spin state ELS can be obtained directly from the energy of a broken-symmetry solution EBS. Then, we arrive to the following expressions for J:
$ {E_{{\rm{HS}}}} - {E_{{\rm{BS}}}} = - 15{J_{{\rm{Mn - Mn}}}} $
(5) $ {E_{{\rm{HS}}}}{\rm{ - }}{E_{{\rm{BS}}}} = - 6{J_{{\rm{Co - Co}}}} $
(6) $ {E_{_{{\rm{HS}}}}} - {E_{{\rm{BS}}}} = - 3{J_{{\rm{Ni - Ni}}}} $
(7) In this case, we intercepted the binuclear fragments of complexes 2~4 and used them for theoretical calcula-tion. Finally, the value of J was obtained. The calculated coupling constant J and related quantities are listed in Table 2. The computed J values (J=-2.41 cm-1 for 2, -12.73 cm-1 for 3 and -14.48 cm-1 for 3) predict antiferromagnetic interaction, and the results are consistent with the experimental data. We have used the non-spin projected, giving a better agreement for complexes 2~4. This is due to the strong localization of the wave function at the metal centers. So, comput-ational techniques produce results in remarkable agreement with the experimental value. The calculation using hybrid B3LYP function to reproduce BS-HS energy gap of complexes is found to not only be successful in describing the magnetic behavior of the complexes correctly in this study, but also yield J that are in excellent agreement with the experimental value.
Table 2
Table 2. Calculated energy and magnetic exchange constant J for complexes 2~4下载:
导出CSV
Complex EBS/a.u. EHS/a.u. Calculated J/cm-1 Experimental J/cm-1 2 -5 198.792 206 -5 198.792 195 -2.41 -0.79 3 -6 201.707 332 -6 201.707 274 -12.73 -8.97 4 -6 830.741 504 -6 830.741 438 -14.48 -11.42 3. Conclusions
In summary, four transition metal coordination polymers have been synthesized by the self-assembly of M(Ⅱ) (M=Mn, Co, Ni) salts with 2, 4-H2oba and auxiliary N-donor ligands. Assemblies of these complexes generate two types of diverse frameworks: two 1D chains and two 2D structures. By comparing of the structures of 1~4, the N-donor ligand and metal salts can also slightly tune the final structural features. Moreover, magnetic properties of 2~4 have also been investigated. According to the crystal structures, the DFT-BS approach was applied to study the magnetic coupling behavior for 2~4, and the result reveals that the calculated exchange coupling constants J were in good agreement with the experimental data.
Supporting information is available at http://www.wjhxxb.cn
-
-
[1]
Wu Y P, Zhou W, Zhao J, et al. Angew. Chem. Int. Ed., 2017, 56:13001-13005 doi: 10.1002/anie.201707238
-
[2]
Huang N, Wang K C, Drake H, et al. J. Am. Chem. Soc., 2018, 140:6383-6390 doi: 10.1021/jacs.8b02710
-
[3]
Shi Z Z, Qin L, Zheng H G. Dalton Trans., 2017, 46:4589-4594 doi: 10.1039/C7DT00614D
-
[4]
Zhai Q G, Bu X H, Zhao X, et al. Acc. Chem. Res., 2017, 50:407-417 doi: 10.1021/acs.accounts.6b00526
-
[5]
Zhang X Y, Li B, Zhang J P. Inorg. Chem., 2016, 55:3378-3383 doi: 10.1021/acs.inorgchem.5b02785
-
[6]
Park J, Xu M, Li F Y, et al. J. Am. Chem. Soc., 2018, 140:5493-5499 doi: 10.1021/jacs.8b01613
-
[7]
Pang J D, Yuan S, Qin J S, et al. Coord. Chem. Rev., 2018, 354:28-45 doi: 10.1016/j.ccr.2017.06.007
-
[8]
Li H Y, Cao L H, Wei Y L, et al. CrystEngComm, 2015, 17:6297-6307 doi: 10.1039/C5CE01008J
-
[9]
黎彧, 邹训重, 顾金忠, 等.无机化学学报, 2018, 34(6):1159-1165 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180620&flag=1LI Yu, ZOU Xun-Zhong, GU Jin-Zhong, et al. Chinese J. Inorg. Chem., 2018, 34(6):1159-1165 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180620&flag=1
-
[10]
银秀菊, 廖蓓玲, 吴汉民, 等.无机化学学报, 2017, 33(6):1043-1050 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20170618&flag=1YIN Xiu-Ju, LIAO Bei-Ling, WU Han-Min, et al. Chinese J. Inorg. Chem., 2017, 33(6):1043-1050 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20170618&flag=1
-
[11]
王记江, 侯向阳, 高楼军, 等.无机化学学报, 2014, 30(7):1616-1620 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20140718&flag=1WANG Ji-Jiang, HOU Xiang-Yang, GAO Lou-Jun, et al. Chinese J. Inorg. Chem., 2014, 30(7):1616-1620 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20140718&flag=1
-
[12]
Tang L, Jing R, Wang J J, et al. Chin. J. Struct. Chem., 2017, 36:1179-1184
-
[13]
Tang L, Fu F, Wang J J, et al. Polyhedron, 2015, 88:116-124 doi: 10.1016/j.poly.2014.12.023
-
[14]
Liu J Q, Wang Y Y, Zhang Y N, et al. Eur. J. Inorg. Chem., 2009:147-154 doi: 10.1002/ejic.200800898
-
[15]
He J H, Xiao D R, Yan S W, et al. Solid State Sci., 2012, 14:1203-1209 doi: 10.1016/j.solidstatesciences.2012.06.004
-
[16]
Xue D X, Lin J B, Zhang J P, et al. CrystEngComm, 2009, 11:183-188 doi: 10.1039/B813226G
-
[17]
Liu J Q, Wang Y Y, Ma L F, et al. CrystEngComm, 2008, 10:1123-1125 doi: 10.1039/b808540b
-
[18]
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 03, Ver.6.1, Gaussian Inc., Wallingford CT, 2004.
-
[19]
Becke A D. J. Chem. Phys., 1997, 107:8554-8560 doi: 10.1063/1.475007
-
[20]
Ruiz E, Rodríguez-Fortea A, Tercero J, et al. Chem. Phys., 2005, 123:074102-074102 http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM16229554
-
[21]
Sheldrick G M. SADABS, A Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 2008.
-
[22]
Sheldrick G M. Acta Crystallogr. Sect. A:Found. Crystallogr., 2015, A71:3-8 https://www.ncbi.nlm.nih.gov/pubmed/25537383
-
[23]
Sheldrick G M. Acta Crystallogr. Sect. C:Cryst. Struct. Commun., 2015, C71:3-8 https://www.ncbi.nlm.nih.gov/pubmed/25567568
-
[24]
Yang W B, Lin X, Blake A J, et al. Inorg. Chem., 2009, 48:11067-11078 doi: 10.1021/ic901429u
-
[25]
Carlin R L. Magnetochemistry. Berlin: Springer, 1986.
-
[26]
Kahn O. Molecular Magnetism. New York: VCH Publishers Inc., 1993.
-
[27]
Ishida T, Kawakami T, Mitsubori S I, et al. J. Chem. Soc. Dalton Trans., 2002:3177-3186 https://pubs.rsc.org/en/content/articlelanding/2002/DT/b202635j#!divAbstract
-
[28]
Beghidja C, Rogez G, Kortus J, et al. J. Am. Chem. Soc., 2006, 128:3140-3141 doi: 10.1021/ja0575023
-
[29]
Ruiz E, Alemany P, Alvarez S, et al. Inorg. Chem., 1997, 36:3683-3688 doi: 10.1021/ic970310r
-
[30]
Zhou X H, Chen Q Q, Liu B L, et al. Dalton Trans., 2017, 46:430-444 doi: 10.1039/C6DT04270H
-
[31]
Rodríguez-Fortea A, Alemany P, Alvarez S, et al. Inorg. Chem., 2001, 40:5868-5877 doi: 10.1021/ic001420s
-
[1]
-
Figure 1 Coordination environment of Mn(Ⅱ) ion of complex 1
Hydrogen atoms and the free H2O molecules are omitted for clarity except the coordination water; 40% ellipsoid probability level; Symmetry codes: #1:-x+1, y, -z+3/2; #2: x, y-1, z
Figure 4 Coordination environment of Mn(Ⅱ) ion in complex 2
All hydrogen atoms are omitted for clarity; Symmetry codes: #1: -x+3/2, y-1/2, -z+3/2
Figure 7 Binuclear Co(Ⅱ)-tetracarboxylate paddle-wheel of 3
Hydrogen atoms are omitted for clarity; Symmetry codes: #1: x, y-1, z; #2:-x, -y+1, -z
Figure 10 Thermal variation of χM and χMT for 2
Open points are the experimental data, and the solid line represents the best fit obtained from the Hamiltonian given in the text
Figure 11 Thermal variation of χM and χMT for 3
Open points are the experimental data, and the solid line represents the best fit obtained from the Hamiltonian given in the text
Figure 12 Thermal variation of χM and χMT for 4
Open points are the experimental data, and the solid line represents the best fit obtained from the Hamiltonian given in the text
Table 1. Crystal data and structural refinement summary of the complexes 1~4
Complex 1 2 3 4 Empirical formula C38H34MnN2O14 C26H16MnN2O5 C21H15CoN2O5 C21H15NiN2O5 Formula weight 797.61 491.35 434.28 434.06 Crystal system Monoclinic Monoclinic Monoclinic Monoclinic Space group P2/c P21/n P21/n P21/n a/nm 1.220 93(17) 0.795 92(7) 1.176 6(3) 1.172 07(12) b/nm 1.156 24(16) 1.546 14(14) 1.033 1(3) 1.020 41(10) c/nm 1.557 72(16) 1.871 30(17) 1.555 5(4) 1.568 53(14) β/(°) 122.053(8) 99.949(2) 103.927(5) 103.921(2) V/nm3 1.863 8(4) 2.268 2(4) 1.835 2(9) 1.820 8(3) Dc/(g·cm-3) 1.421 1.439 1.572 1.583 Z 2 4 4 4 μ/mm-1 0.426 0.623 0.973 1.103 Reflection collected, unique 9 116, 3 286 (Rint=0.020 1) 11 259, 4 006 (Rint=0.052 2) 8 969, 3 231 (Rint=0.033 8) 11 153, 4 408 (Rint=0.035 5) Data, restraint, parameter 3 286, 3, 258 4 006, 0, 307 3 231, 0, 262 4 408, 0, 262 Goodness-of-fit (GOF) on F2 1.050 1.030 1.015 1.001 Final R indices [I>2σ(I)] R1=0.036 3, wR2=0.104 2 R1=0.043 3, wR2=0.069 9 R1=0.035 4, wR2=0.086 0 R1=0.039 8, wR2=0.080 8 Largest difference in peak and hole/(e·nm-3) 474 and -209 199 and -200 354 and -264 428 and -256  下载: 导出CSV
下载: 导出CSV
Table 2. Calculated energy and magnetic exchange constant J for complexes 2~4
Complex EBS/a.u. EHS/a.u. Calculated J/cm-1 Experimental J/cm-1 2 -5 198.792 206 -5 198.792 195 -2.41 -0.79 3 -6 201.707 332 -6 201.707 274 -12.73 -8.97 4 -6 830.741 504 -6 830.741 438 -14.48 -11.42
下载: 导出CSV
-













 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 1136
- HTML全文浏览量: 118
