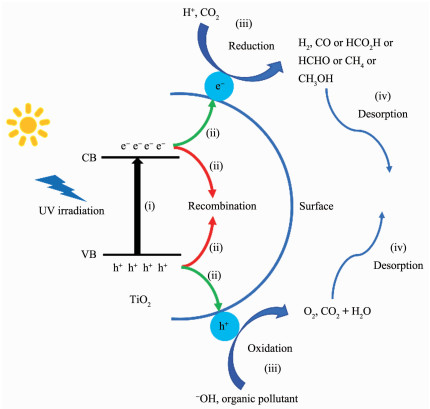
图 1.
TiO2的光催化机理图
Figure 1.
Photocatalytic mechanism diagram of TiO2

目前,环境污染和资源短缺是亟需解决的两大全球性问题。光催化技术,源于对植物光合作用的人工模拟,可以实现太阳能向化学能的转化,被认为是一种绿色高效的能源转换模式。自1972年日本科学家Fujishima和Honda发现TiO2在光电驱动下的水分解现象之后[1],TiO2便得到不少研究者的青睐,随后在降解污染物[2-3]、水分解析氢产氧[4-6]和CO2还原[7-8]等领域上均表现出潜在的应用价值。TiO2具有廉价、无毒、化学性质稳定等优异的性质,但受限于较宽的能带间隙和快速的光生电子-空穴对复合,难以满足实际应用的要求。
针对以上问题,研究者们提出了元素掺杂、贵金属表面沉积和异质结等改性方法,以致力于改善TiO2的2个缺陷。其中,选择与TiO2的能带结构相匹配且能带间隙小的半导体,与TiO2构建异质结,表现出更高的量子产率和更宽的光谱响应范围。在异质结中,能有效提高TiO2光催化性能的主要包含2类:type-Ⅱ异质结和Z型异质结。type-Ⅱ异质结的2种半导体的能带交错排列,光激发电子跃迁和迁移导致电子和空穴在不同半导体上累积,从而实现电子与空穴的分离。然而,这种电子迁移途径会弱化type-Ⅱ异质结的氧化还原能力。Z型异质结具有与type-Ⅱ异质结相同的能带排列,但电子传递的途径不同,电子在半导体间的迁移路径类似于英文字母“Z”,因此称之为Z型光催化体系。这种独特的电子迁移途径导致Z型异质结在增加电子与空穴分离效率的同时,仍然能保持较高的氧化还原能力[9]。本文以TiO2基Z型异质结光催化剂为主题,介绍了TiO2光催化剂、异质结光催化剂和TiO2基Z型异质结光催化剂的能带结构与电子迁移原则,对比了type-Ⅱ异质结与Z型异质结的异同点,讨论了Z型异质结的优势所在, 并在此基础上提出了区分type-Ⅱ异质结与Z型异质结的3种方法。另外,归纳整理了TiO2基Z型异质结在光催化领域内的优秀研究成果,展现出TiO2基Z型异质结在污染物降解、水分解以及CO2还原等领域上广阔的应用前景。在此基础上,我们希望能借此拓宽TiO2在光催化领域中的应用,并为研究者们提供构建新型光催化剂的一些启发和建议。
TiO2是一种能带间隙为3.0~3.2 eV的半导体,在自然界中存在3种形式的晶型:锐钛矿、金红石和板钛矿。晶体结构的差异会导致TiO2的能带间隙和光催化活性发生变化。锐钛矿的能带间隙为3.2 eV,光催化活性最高;金红石的能带间隙为3.0 eV,光催化活性次之;板钛矿不稳定,不宜作为光催化剂。TiO2的光催化过程一般包含4步(图 1):(ⅰ)TiO2能带较宽,仅受紫外光激发,电子(e-)从价带(VB)跃迁到导带(CB)上,并在导带上留下带正电的空穴(h+);(ⅱ)电子与空穴分离并扩散到TiO2表面,或在体相内复合而湮灭;(ⅲ)电子和空穴与吸附在TiO2表面的反应物发生氧化还原反应,例如H+和CO2的还原、OH-和有机污染物的氧化等;(ⅳ)产物从TiO2表面解吸。通过时间分辨吸收光谱研究光生载流子在TiO2中形成、扩散、复合以及传递的反应动力学(图 2)[10]。结果显示,各光催化反应的时间十分短暂,其中载流子在TiO2表面上复合的时间为1~10 ps,在体相内的复合时间大于20 ns,如此快速的复合并不利于其它光催化反应的进行。因此,提高TiO2光催化性能的关键在于能带调控和电子-空穴分离,其中涉及到一些常用的方法包括:元素(C[11]、S[12]、N[13]、I[14]、Fe[15]、Ti[16]、Bi[17]等)掺杂,或贵金属(Au[18]、Pt[19]、Ag[20]、Pd[21-22]等)表面沉积,或引入窄带隙的半导体(NiO[23]、CuS[24]、NiS[25]、MoS2[26]、g-C3N4[27]、WO3[28]、Ag2O[29]、SnS2[30]等)作为光敏组分与之构建异质结等。前2种方法均存在一定的局限性:一是元素掺杂所形成的杂质能级有可能成为电子与空穴的复合中心;二是贵金属价格昂贵且在催化反应过程中易流失。相较之下,异质结是一种更理想的光催化剂。
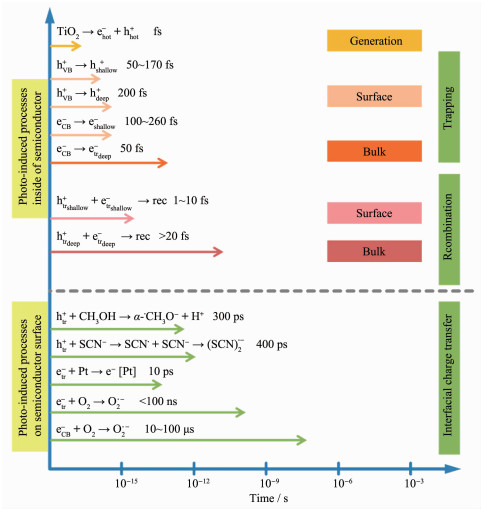
ehot- and hhot+ are excited electrons and holes, respectively; eCB- and hCB+ are electrons in conduction band and holes in valence band, respectively; eshallow-, hshallow+, edeep- and hdeep+: are electrons and holes in shallow or deep trap, respectively; etr- and htr+ are trapped electrons and holes, including etrshallow-, htrshallow+, etrdeep- and htrdeep+; rec is short for recombination
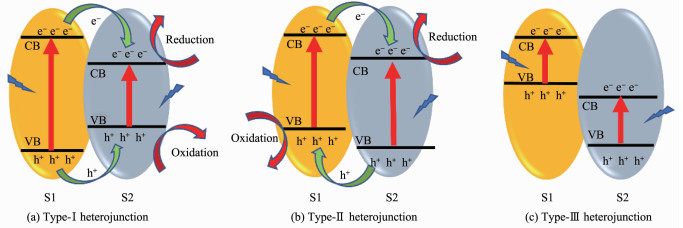
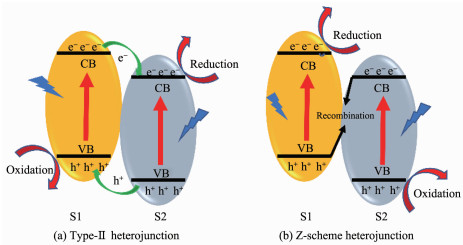
异质结,是指2种能带结构不同的半导体相接触的界面[31]。根据半导体的能带排列不同,异质结可以分为type-Ⅰ异质结、type-Ⅱ异质结和type-Ⅲ异质结,它们的能带排列如图 3[32]所示。图 3a为type-Ⅰ异质结,2种半导体(S1和S2)的能带呈现跨越式排列。由于电子易迁移到导带能级较低的半导体上而空穴易迁移到价带能级较高的半导体上,导致电子与空穴均迁移到半导体2上,电子与空穴的分离效率低。图 3b为type-Ⅱ异质结,2种半导体的能带呈现交错式排列。电子从半导体1的导带上迁移到半导体2的导带上,电子在半导体2上累积;空穴从半导体2的价带上迁移到半导体1的价带上,空穴在半导体1上累积,最终实现了电子与空穴在空间上的有效分离。图 3c为type-Ⅲ异质结,2种半导体的能带呈现中断式排列,电子与空穴在界面间无迁移行为。综合以上3种异质结,type-Ⅱ异质结是这3种异质结中提高电子-空穴对空间分离最有效的传统异质结[33]。
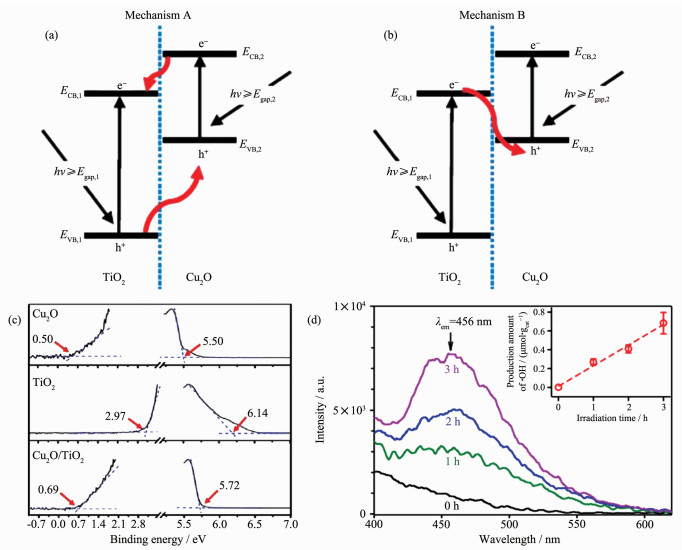
除了以上3种传统异质结外,TiO2-石墨烯异质结[34-36]、p-n结[23, 37-38]和Z型异质结[25, 39-40]也具有分离电子与空穴的能力,且在电荷传递速度或氧化还原能力上优于type-Ⅱ异质结。其中,Z型异质结作为一种新型异质结,在能带的排列上与type-Ⅱ异质结十分相似,均呈现为交叉式排列,而两者的不同点在于电子的迁移方向不同(图 4)。图 4a为type-Ⅱ异质结的能带排列与电子迁移机理,电子与空穴分别在半导体2的导带和半导体1的价带上累积,实现了在空间上的分离。然而,这种迁移方式削弱了光催化剂的氧化还原能力,即电子迁移到能级较低的导带处,还原能力减弱;空穴迁移到能级较高的价带处,氧化能力减弱。图 4b为Z型异质结的能带排列与电子迁移机理,半导体2导带上的电子与半导体1价带上的空穴复合而湮灭,残留的电子主要存在于半导体1的导带上而空穴主要存在于半导体2的价带上,同样也实现了电子与空穴在空间上的分离。然而,相对于type-Ⅱ异质结,Z型异质结的优势在于电子在较高的能级上累积而空穴在较低的能级上累积,氧化还原能力更强。Grela等[41]报道了Cu2O/TiO2复合物以Z型电子传递的方式,实现了CO2的还原以及阻止了Cu2O的光腐蚀效应。根据紫外光电子能谱图(图 5c)计算Cu2O的导带(ECB, 2)和价带(EVB, 2)位置分别为-1.27和0.76 eV,而TiO2的导带(ECB, 1)和价带(EVB, 1)位置分别为-0.57和2.59 eV。按照图 5a的type-Ⅱ异质结的催化机理(其中Egap, 1和Egap, 2分别为TiO2和Cu2O的带隙),还原反应发生在TiO2的导带上,氧化反应发生在Cu2O的价带上,而水氧化产生羟基自由基(·OH)的氧化还原电势为2.27 eV,Cu2O价带上的空穴不足以氧化水产生·OH。然而,通过7-羟基香豆素的荧光光谱(图 5d)定量探测·OH,结果证实了·OH的存在。因此,可推测复合物并非type-Ⅱ异质结而是Z型异质结,氧化能力更强,水氧化反应发生在TiO2的价带上(图 5b)。由这一案例可知,Z型异质结在氧化还原能力上优于type-Ⅱ异质结。
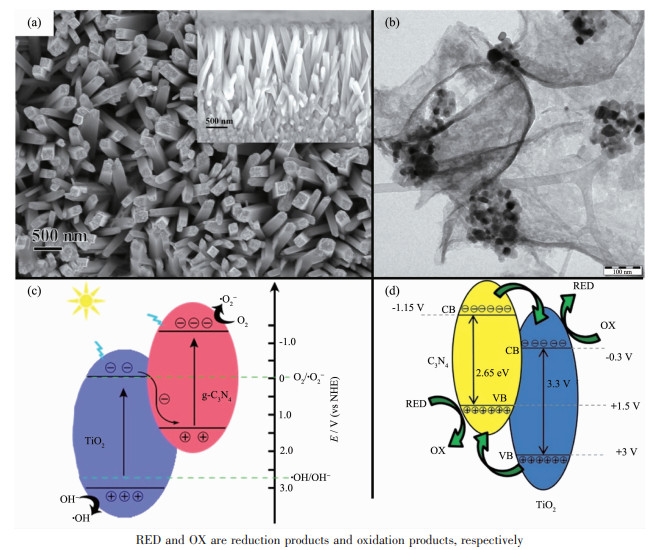
根据上述讨论,在构建异质结时更倾向于得到Z型结构。然而,当复合物中2种半导体的能带呈现交错式排列时,很难去分辨复合物是属于type-Ⅱ异质结还是Z型异质结。例如:在TiO2/g-C3N4纳米复合物的研究中,Li等[42]采用简单的饱和溶液法合成了TiO2@g-C3N4核壳纳米棒阵列(图 6a),并通过第一性原理的计算和模拟,推测出TiO2导带与g-C3N4价带的能级差为1.34 eV,小于TiO2导带与g-C3N4导带的能级差1.44 eV,电子更容易从TiO2导带向g-C3N4价带迁移。因此,TiO2@g-C3N4核壳纳米棒阵列符合Z型光催化机理,如图 6c所示。另外,Yu等[43]通过系统的理论计算,验证了TiO2和g-C3N4之间的界面相互作用促进内建电场的形成,从而加速电子从TiO2向g-C3N4的转移,这也印证了TiO2/g-C3N4属于Z型异质结的光催化机理。然而,在Martin等[44]的研究中,合成的具有相同化学结构的TiO2/g-C3N4纳米复合物(图 6b)却被认定为type-Ⅱ异质结(图 6d)。表 1统计了现有研究中所报道的一系列TiO2/g-C3N4纳米复合物。由表可知,g-C3N4和TiO2半导体复合时,有可能得到Z型异质结,也有可能得到type-Ⅱ异质结,至于造成这种结果的原因依然尚不明确。由于无法预知复合材料中异质结的种类,研究者常通过后续的测试和表征来区分它们,而区分的依据是3.1中所讨论的2种异质结的差异:(1)电子传递的方向不同;(2)发生氧化还原的能带位置不同。目前,用来区分type-Ⅱ异质结和Z型异质结的方法和测试包括:依靠氧化还原反应的电势、能带弯曲的方向和XPS偏移的方向来区分。
 下载:
导出CSV
下载:
导出CSV
| Photocatalyst | Type of heterojunction | Application | Reference |
| TiO2@g-C3N4 core-shell nanorods arrays | Z-scheme | Degradation of RhB | [42] |
| TiO2g-C3N4 | Type-Ⅱ | Reduction of CO2 and decomposition of N2O | [44] |
| Hierarchical TiO2/g-C3N4 hybrids | Z-scheme | Degradation of RhB | [45] |
| TiO2/g-C3N4 | Type-Ⅱ | Degradation of MO | [46] |
| Mesoporous TiO2/g-C3N4 microspheres | Type-Ⅱ | Degradation of phenol | [47] |
| Spherical TiO2/g-C3N4 | Type-Ⅱ | Decomposition of MB | [48] |
| TiO2/g-C3N4 nanofibers | Z-scheme | Degradation of RhB | [49] |
| Nanofibrous TiO2/g-C3N4 | Z-scheme | Degradation of RhB | [50] |
| Ultrathin g-C3N4/anatase TiO2 nanosheets | Type-Ⅱ | Degradation of MB | [51] |
| Core-shell TiO2@g-C3N4 hollow microspheres | Type-Ⅱ | Degradation of RhB | [52] |
| TiO2/g-C3N4 | Z-scheme | Decomposition of N2O | [53] |
| TiO2@g-C3N4 core-shell quantum heterojunction | Type-Ⅱ | Degradation of tetracycline | [54] |
| TiO2/C3N4 | Z-scheme | Water splitting | [55] |
| g-C3N4-TiO2 | Z-scheme | Remove of propylene | [56] |
| g-C3N4-TiO2 | Z-scheme | Decomposition of formaldehyde | [57] |
| RhB: rhodamine B; MB: methylene blue; MO: methyl orange | |||
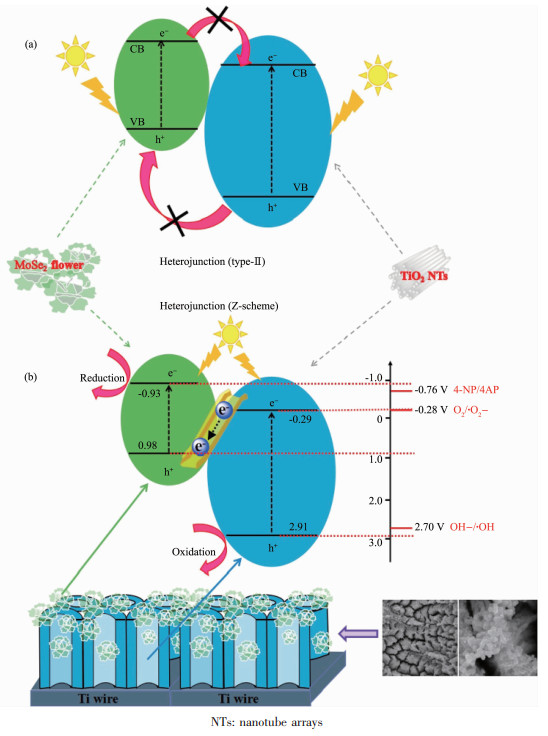
Z型异质结的氧化还原能力大于type-Ⅱ异质结,与之相匹配的氧化还原反应的电势范围更宽。当反应物的氧化还原反电势处在2个半导体的导带或价带之间,通过检测该氧化还原反应产物的存在可以判断异质结的种类:若产物存在,则光催化剂为Z型异质结。Yang等[58]选择MoSe2和TiO2作为构建异质结的2种半导体。MoSe2的导带和价带位置分别为-0.93和0.98 V;TiO2的导带和价带位置分别为-0.29和2.91 V。跟据type-Ⅱ异质结的电荷传递方向(图 7a),能发生氧化还原反应的电势范围应在-0.29~0.98 V之间,而Z型异质结的电荷传递方向不同(图 7b),能发生氧化还原反应的电势范围应在-0.93~2.91 V。在光催化反应中,通过紫外-可见吸收光谱和以二甲基吡啶氮氧化物(DMPO)作为自由基捕获剂的电子自旋共振谱图,检测到对硝基苯酚(4-NP)还原为对氨基苯酚(4-AP)以及超氧自由基(·O2)和·OH的存在。其中4-NP/4-AP的标准电极电势为-0.76 V,OH-/·OH的标准电极电势为2.70 V。type-Ⅱ异质结不足以还原4-NP和氧化OH-,因此可以判断MoSe2和TiO2复合形成了Z型异质结。这种方法是目前研究中最常用于确定异质结种类的方法,而其中检测氧化还原反应和产物的手段较多,包括紫外-可见吸收光谱[58]、荧光光谱[56, 59]、自由基捕获实验[60]以及电子自旋共振谱图[58, 61-63]。但这种方法存在一定局限性:(1)氧化还原反应的电势必须处在2个半导体的导带或价带之间;(2)产物不存在时,无法判断异质结的种类。
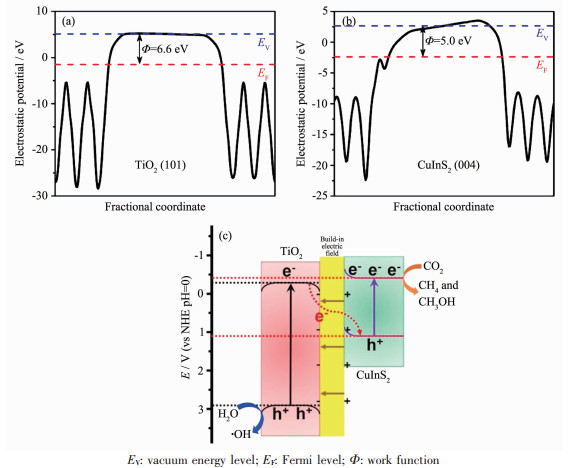
能带弯曲是半导体材料在复合过程中常见的一种现象。半导体费米能级(EF)的差异导致电荷在半导体间的转移,最终形成反方向的内建电场以及能带弯曲[64]。能带弯曲和内建电场会加速电子沿着电场的方向转移,同时也会抑制反方向的电子转移。Yu等[65]通过密度泛函理论(DFT)计算得到TiO2(101)面和CuInS2(004)面的功函数(Φ)分别为6.6和5.0 eV,TiO2的费米能级低于CuInS2的费米能级(图 8(a,b))。因此当TiO2与CuInS2相接触时,为平衡费米能级,电子从CuInS2向TiO2转移而在靠近TiO2处留下带负电区域,空穴从TiO2向CuInS2转移而在靠近CuInS2处留下带正电区域,同时TiO2的能带向下弯曲,CuInS2的能带向上弯曲(图 8c)。最终,电子只能从TiO2导带转移到CuInS2价带上与空穴复合,从而构建出一种Z型光催化系统。费米能级与半导体的温度、元素掺杂以及电子结构有关[66],理论计算得到的费米能级与实际结果仍然存在一定的偏差,因此通过能带弯曲的方向去区分Z型异质结和type-Ⅱ异质结也存在一定的缺陷。
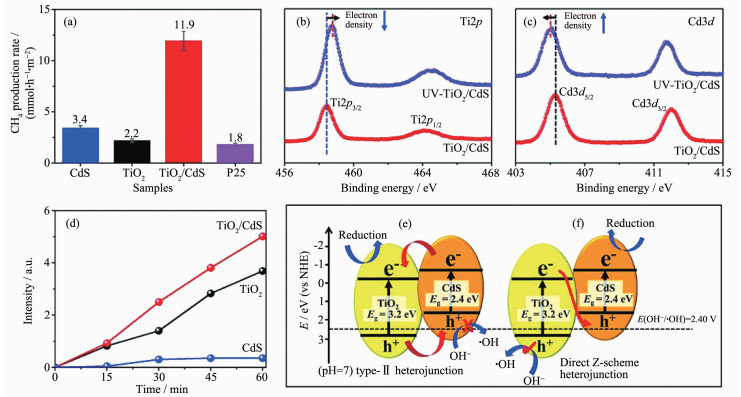
异质结受光激发,电子在2种半导体间传递,最终导致一个半导体电子云密度的下降而另一个半导体电子云密度的升高,在X射线光电子能谱(XPS)中表现为特征峰的偏移。Z型异质结和type-Ⅱ异质结的电子迁移方向不同,对应在XPS中特征峰偏移的方向相反,因此通过XPS偏移方向也可以区分异质结的种类。Yu等[67]报道了一种原位辐射XPS方法,用于对直接Z型异质结TiO2/CdS的研究。365 nm的紫外光照射TiO2/CdS后,在Ti2p高分辨XPS谱图(图 9b)中,特征峰向高能区偏移,证明了Ti原子周围电子云密度的降低;在Cd3d高分辨XPS谱图(图 9c)中,特征峰向低能区偏移,证明了Cd原子周围电子云密度的增加。结合两者推测出TiO2/CdS复合物符合Z型光催化体系(图 9f)。同时根据7-羟基香豆素的荧光光谱探测到复合物相较于单一组分易产生更多的·OH(图 9d),而按照type-Ⅱ异质结的光催化机理(图 9e),复合物不足以氧化OH-产生·OH,进一步地证明了TiO2/CdS复合物是一种Z型异质结。由于Z型异质结赋予了复合物更强的氧化还原能力,所制备的TiO2/CdS复合薄膜对CO2的还原反应表现出高的催化活性,即CH4的转化率达到11.9 mmol·h-1·m-2(图 9a)。XPS图谱中特征峰的偏移最直接的原因是电子云密度的变化,而造成电子云密度变化的原因不仅仅是电子的转移。因此在利用XPS分辨异质结种类时,还需要结合其它方法加一验证。
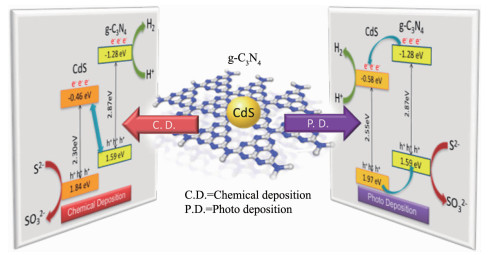
在上述的讨论中,研究者大多先合成材料,然后通过材料性质的表征来确定异质结的类型。Sun等[68]提供一种新的思路,通过改变制备方法有目的地得到了type-Ⅱ异质结或Z型异质结CdS/g-C3N4,如图 10所示。利用化学沉积法在g-C3N4上自由地沉积CdS纳米颗粒,得到的是Z型异质结,而利用光沉积法将CdS纳米颗粒有选择性地沉积在g-C3N4的电子传递位点上,导致光生电子趋向于从g-C3N4转移到CdS上,最终得到的是type-Ⅱ异质结。与此类似,我们发现构建的TiO2/CdS复合物的异质结种类与CdS在TiO2上的沉积方式也存在上述这种关系。譬如,离子层吸附和化学还原法是一种将CdS沉积在TiO2上的常用方法[67, 69],这是一种典型的化学沉积方法,通过这种方法构建出的是Z型异质结;而光沉积法则利用紫外光激发的TiO2所具有的强还原性,将S8还原为S2-,继而将反应体系中的Cd2+转化为CdS而沉积在TiO2上[70-71],最终得到了type-Ⅱ异质结。
另外,在最近的研究中,Dong等[72]发现TiO2/CdS复合物的异质结种类与CdS沉积在TiO2上的晶面也存在一定的关系。将TiO2选择性地沉积在TiO2纳米片的(001)晶面上,形成了type-Ⅱ异质结;而将TiO2选择性地沉积在TiO2纳米片的(101)晶面上,形成了Z型异质结。TiO2的(001)晶面(0.90 J·m-2)比(101)晶面(0.44 J·m-2)具有更高的能量密度[73],导致电子在(101)面上富集而空穴在(001)面上富集。因此,(101)晶面作为一种电子供体易向CdS转移电子,从而形成Z型异质结;(001)晶面作为一种电子受体易接收来自CdS导带上的电子,从而形成type-Ⅱ异质结。受此启发,TiO2的2种晶面同时存在时,分别来自这2种晶面的空穴和电子也会复合,从而形成一种Z型异质结。例如,Lv等[74]以暴露(001)高能晶面的TiO2修饰TiO2纳米纤维,形成了Z型光催化反应机制。综合而言,以上研究有目的地构建TiO2基Z型异质结提供了一些新思路,但它是否具有普适性,我们还尚未可知,需要进一步地验证。
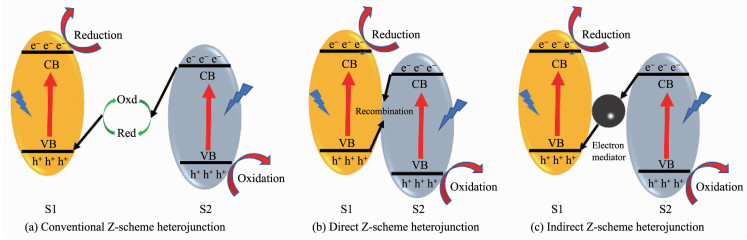
Z型异质结的概念最早是在1979年由Bard等在模拟植物光合作用的基础上提出[75]。这种传统的Z型异质结(图 11a)由2种没有物理接触的半导体以及氧化还原(Oxd/Red)介质组成,其中氧化和还原介质分别是电子的受体和供体。常用的氧化还原介质包括:IO3-/I-和Fe3+/Fe2+ [76]。电子在传统Z型异质结中的转移过程为:来自半导体2上的光生电子还原氧化介质,还原介质接收来自半导体1上的空穴而被氧化,最终导致电子在半导体1导带上累积而空穴在半导体2价带上累计,实现了电子与空穴在空间上的分离。对于这种传统的Z型光催化体系,其受限于液相氧化还原介质的影响而难以适应更复杂的光催化反应环境。
2006年,Tada等[77]基于对CdS-Au-TiO2三组分光催化剂的研究,第一次提出了全固态Z型异质结的概念。前期,全固态Z型异质结主要是三元体系的间接Z型异质结(图 11c),包含在2种半导体和夹杂在中间的电子媒介,电子媒介一般由石墨烯[78-79]或贵金属(Au[77, 80]、Ag[81]、Pt[82]等)组成。石墨烯具有高的电荷迁移率,贵金属具有大的功函数,两者都易接受来自半导体导带上的电子。正是由于这种特性,固态电子媒介会促进导带电势较低的半导体上的光诱导电子与价带电势较高的另一半导体上的空穴结合,而氧化还原能力较强的电子和空穴仍保留在2个半导体上。与传统Z型异质结类似,最终也形成了电子与空穴在空间上的分离,这种分离给催化剂带来了更高的催化活性和氧化还原能力。譬如,Liu等[81]在Tada的基础上构建了一种基于CdS纳米线的三元CdS-Au-TiO2的Z型异质结。相对于CdS-TiO2和纯的CdS,这种Z型异质结光催化剂展现出较快的析氢速率(1 910 μmol·g-1·h-1),同时也抑制了CdS的光腐蚀。另外,Li等[83]构建的g-C3N4/Pd/TiO2和Liu[78]等构建的g-C3N4-RGO-TiO2间接Z型异质结均表现出优于单组分和双组分的催化活性。
在间接Z型异质结的基础上,Yu等[57]于2013年首次提出了直接Z型异质结(图 11b)的概念。直接Z型异质结是二元体系,不包含电子媒介,2种半导体直接接触,电子直接在2种半导体的界面间传递,也实现了电子与空穴在空间上的分离。半导体间的固-固接触界面的性质决定了电子传递的阻力,而阻力的大小与接触界面的形成方式有很大关系[39]。因此,固-固接触界面的设计对于直接Z型异质结的构建具有重要意义。理论分析认为,由于界面缺陷可以作为电荷俘获的中心,固-固接触界面上的大量表面缺陷可以起到欧姆接触的作用[84]。氧空位是金属氧基半导体的一种固有缺陷,对调控界面状态和捕获半导体间的电荷具有重要的意义[85]。Yue等[86]通过液氮淬火法制备了表面富含氧空位的TiO2中空球,限制了光生载流子的复合以及提高了TiO2对可见光的吸收,催化剂的产氢量在3 h可见光照射下达到1 240.5 μmol。Zhang等[39]在直接Z型三元硼碳氮化合物(BCN)-TiO2中,通过NaBH4还原形成界面氧空位层,促进了载流子在界面处的传递,展现出良好的可见光析氢速率(3.01 μmol·h-1),约为纯TiO2的7倍和纯BCN的11倍。
TiO2基Z型异质结能有效地提高TiO2的光催化性能和对可见光的利用率,因此可以拓展TiO2在光催化领域上的应用。目前,光催化领域的研究热点主要包括:污染物降解、水分解和CO2还原。合理地构建TiO2基Z型异质结,提高材料的光催化活性,可以有效地促进理论研究向实际应用的转化,最终解决环境污染和能源危机两大全球性问题。本文归纳整理了部分TiO2基Z型异质结在光催化领域中的应用(表 2),并进一步讨论TiO2基Z型异质结在污染物降解、水分解和CO2还原上的应用实例和研究进展。
下载:
导出CSV
| Photocatalyst | Light source | Application | Photocatalytic efficiency | Reference |
| TiO2@g-C3N4 core-shell nanorod arrays | 100 W Xe lamp | Degradation of RhB | 180 min, 95.68% | [42] |
| Hierarchical TiO2/g-C3N4 | 350 W Xe arc lamp | Degradation of RhB | 0.055 min-1 | [45] |
| TiO2/g-C3N4 nanofibers | 300 W Xe lamp | Degradation of RhB | 70 min, 99% | [49] |
| Nanofibrous TiO2/g-C3N4 | 500 W Xe lamp | Degradation of RhB | 0.053 45 min-1 | [50] |
| TiO2/g-C3N4 | 8 W Hg lamp | Photocatalytic N2O | 0.001 1 min-1 | [53] |
| BiOI/TiO2 | 500 W Xe lamp | Degradation of MO | 25 min, 85.0% | [87] |
| g-C3N4-Ti3+/TiO2 nanotube arrays | 11 W incandescent lamp | Degradation of phenol | 7 h, 74% | [88] |
| g-C3N4-RGO-TiO2 | 300 W Xe lamp | Degradation of MB | 0.013 7 min-1 | [78] |
| Rutile TiO2/g-C3N4 quantum dots | 500 W Xe lamp | Degradation of RhB | 0.011 5 min-1 | [61] |
| g-C3N4 nanosheets/TiO2 nanosheets | 300 W Xe lamp | Degradation of MO | 0.037 31 min-1 | [89] |
| TiO2 nanotubes/Ag3PO4 | Sunlight irradiation | Degradation of MB | 0.048 min-1 | [90] |
| Nitrogen-doped carbon dots/{001} TiO2 nanosheets |
350 W Xe lamp | Degradation of diclofenac | 60 min, 91.5% | [91] |
| Carbon-modified TiO2/WO3 nanofibers | 300 W Xe arc lamp | Hydrogen production | ~1 570 μmol·g-1·h-1 for H2 | [92] |
| TiO2/WO3/Pt | 300 W Xe arc lamp | Hydrogen production | 128.66 μmol·g-1·h-1 | [82] |
| Anatase/rutile TiO2 nanofibers | 350 W Xe arc lamp | Hydrogen production | 6 480 μmol·g-1·h-1 | [93] |
| WO3-x quantum dots/TiO2 | 300 W Xe lamp | Hydrogen production | 17 700 μmol·g-1·h-1 | [94] |
| TiO2/WO3/Au nanofibers | 300 W Xe arc lamp | Hydrogen production | 5 393 μmol·g-1·h-1 | [95] |
| CuZn-TiO2 | solar light | Hydrogen production | 14 521 μmol·g-1·h-1 | [96] |
| Au/Pt/WO3/TiO2 nanofibers | 300 W Xe arc lamp | Hydrogen production | 242.09 μmol·g-1·h-1 | [97] |
| g-C3N4/TiO2 | 300 W Xe lamp | Hydrogen production | 12 600 μmol·g-1·h-1 for H2 | [59] |
| TiO2/C3N4 | 150 W Xe lamp | Water splitting | 251 μmol·g-1·h-1 for H2 | [55] |
| 121.5 μmol·g-1·h-1 for O2 | ||||
| TiO2/Cu2O | 1 kW Hg (Xe) arc lamp | Reduction of CO2 | 2.11 μmol·g-1·h-1 for CO | [41] |
| Cu2O/TiO2 | 300 W Xe lamp | Photocatalytic propane | 0.54 min-1 for C3H6 | [98] |
| TiO2/CuInS2 | 350 W Xe arc lamp | Reduction of CO2 | 2.5 μmol·g-1·h-1 for CH | [65] |
| 0.86 μmol·g-1·h-1 for CH3OH | ||||
| ZnIn2S4/TiO2 | 300 W Xe lamp | Reduction of CO2 | 1.135 μmol·g-1·h-1 for CH4 | [99] |
目前,研究报道的污染物主要包括水溶性染料[2, 3, 100]、重金属离子[101-103]、苯酚类[47, 88, 104]、抗生素药物[54]以及一些有毒有害的气体[44, 53, 105]。光催化是一种能有效降解污染物的绿色且经济的方法,光催化降解污染物的过程一般包含以下10个反应:
|
${\rm{photocatalyst}} + h\nu \to {{\rm{e}}_{{\rm{CB}}}}^ - + {{\rm{h}}_{{\rm{VB}}}}^ + $ |
(1) |
|
${{\rm{e}}_{{\rm{CB}}}}^ - + {{\rm{O}}_2} \to \cdot {{\rm{O}}_2}^ - $ |
(2) |
|
${{\rm{h}}_{{\rm{VB}}}}^ + + {{\rm{H}}_2}{\rm{O}} \to \cdot {\rm{OH + }}{{\rm{H}}^ + } $ |
(3) |
|
${{\rm{h}}_{{\rm{VB}}}}^ + + {\rm{O}}{{\rm{H}}^ - } \to \cdot {\rm{OH}} $ |
(4) |
|
$ \cdot {{\rm{O}}_2}^ - + {{\rm{H}}^ + } \to {\rm{HOO}} \cdot $ |
(5) |
|
${\rm{HOO}} \cdot {{\rm{e}}_{{\rm{CB}}}}^ - \to {\rm{HO}}{{\rm{O}}^ - } $ |
(6) |
|
${\rm{HO}}{{\rm{O}}^ - } + {{\rm{H}}^ + } \to {{\rm{H}}_{\rm{2}}}{{\rm{O}}_{\rm{2}}} $ |
(7) |
|
${{\rm{H}}_2}{{\rm{O}}_2} + h\nu \to 2 \cdot {\rm{OH}} $ |
(8) |
|
${{\rm{e}}_{{\rm{CB}}}}^ - + {{\rm{h}}_{{\rm{VB}}}}^ + \to {\rm{heat\;or\;other\;energy}} $ |
(9) |
|
$\begin{array}{l} {{\rm{h}}_{{\rm{VB}}}}^ + , \cdot {{\rm{O}}_2}^ - {\rm{or}} \cdot {\rm{OH + pollutants}} \to \\ {\rm{\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;\;C}}{{\rm{O}}_2}, {{\rm{H}}_{\rm{2}}}{\rm{O\;or\;other\;products}} \end{array} $ |
(10) |
在上述反应中,空穴(hVB+)、·O2-和·OH是3种主要的并可用于降解污染物的活性物质,其中·OH的氧化能力强(标准氧化还原电势为+2.8 V),可以氧化大部分偶氮染料[100]。反应(10)为污染物的降解反应,3种活性物质均参与其中,且在降解反应中有着不同的贡献程度,通常通过自由基的捕获实验可以判断各活性物质对降解反应的贡献程度。每一种活性物质都有相应的捕获剂:空穴的捕获剂有乙二醇胺(TEOA)和草酸铵(AO),·O2-的捕获剂有苯醌(BQ)和抗坏血酸,·OH的捕获剂有异丙醇(IPA)。反应(9)为电子-空穴对的复合,该反应的发生会减小电子和空穴的浓度,不利于反应物种的产生。反应机理可以印证,提高光催化剂催化活性的关键因素在于减小电子与空穴的复合几率。根据以上结论,TiO2基Z型异质结将电子与空穴分离在不同的半导体上,实现了电子与空穴在空间上的分离,进而提高光催化降解污染物的效果。
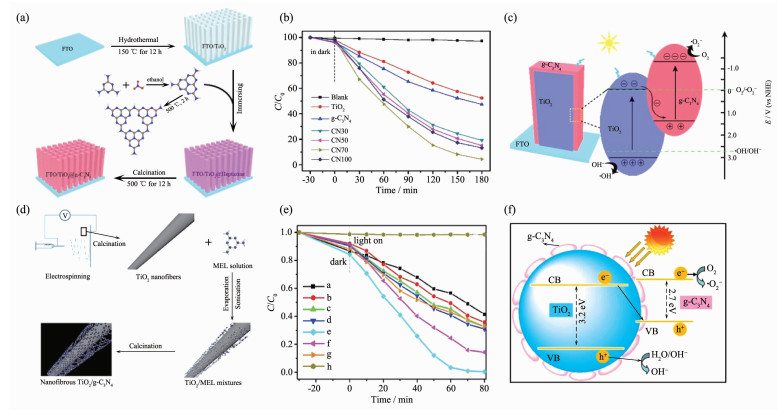
Li等[42]通过三步热沉积法(图 12a)制备了TiO2 @g-C3N4核壳纳米棒阵列,并通过调控反应溶液中三聚氰胺(MEL)饱和溶液和去离子水的体积比,得到了一系列g-C3N4含量不同的复合物,包括CN30、CN50、CN70和CN100(其中30、50、70、100分别代表MEL饱和溶液占去离子水体积的30%、50%、70%、100%)。图 12c为污染物降解的机理图,Li等在DFT的理论基础上进行了第一性原理的模拟,验证了其机理符合Z型异质结机理。通过对RhB的光催化降解实验的结果(图 12b)分析,发现在3 h的可见光照射下,CN70样品对RhB的降解效果基本可以达到100%。另外,Wang等[50]以MEL为原料,通过静电纺丝和煅烧处理得到了一系列纳米纤维状的TiO2/g-C3N4异质结光催化剂,其中TiO2在复合物中的质量分数为0、5.10%、11.57%、20.54%、29.30%、40.36%和100%,分别对应着图 12e中的a~f,h为空白对照实验。g-C3N4包裹TiO2纳米纤维形成一种核壳结构(图 12d),这种光催化剂同样也被推测为Z型异质结(图 12f)。在人工模拟的太阳光照射下,TiO2质量百分数为29.30%的复合材料对RhB的降解效果最佳,即80 min内降解效率达到100%。从这2种材料的结构与降解效果来看,无论是TiO2@g-C3N4核壳纳米棒阵列还是TiO2/g-C3N4异质结光催化剂,都被认定为Z型异质结,并且在可见光下对RhB都有较好的降解效果,这一结果也验证了Z型异质结对材料的光催化性能起积极作用。
目前,TiO2基Z型异质结光催化剂降解污染物的理论研究逐渐发展,在污染物降解上的应用也取得一定的进展,但要将理论延伸到应用中还需要克服许多技术难点,其中包括应付环境的复杂性,克服催化剂回收难的问题,提高催化剂的稳定性以及提高光能的利用率等。Wu等[106]利用蒙脱土(MMT)为载体,接枝纤维素为模板,低温合成了cellulose-g-poly(4-vinylpyridine)/MMT/TiO2复合材料,即提高催化剂的吸附能力又解决了催化剂难回收的问题。同时,现阶段也缺乏考察催化剂催化活性的统一评价标准,这主要是因为试验过程中的影响因素较多,且很难保持一致,其中包括光源的辐射强度、温度、pH、催化剂的用量等等。
近年来,光催化技术在水分解领域中的应用引起了研究者们的广泛关注。2014年,Martin等[107]以尿素为前驱体煅烧得到了g-C3N4,在光催化分解水的实验中展现出高达26.5%的量子产率和高的制氢速率(全光谱照射下制氢速率为20 000 μmol·g-1·h-1,可见光照射下制氢速率为3 300 μmol·g-1·h-1)。利用光催化技术,可以创造一种理想的循环体系,即水分解产生氢气,氢气燃烧产生的水又可以进一步分解为氢气,而完成这一循环的关键是寻找一种合适的光催化剂。光催化水分解包含2个半反应:(1) H+从光催化剂的导带上得到电子,被还原生成H2;(2) H2O分子被光催化剂价带夺取电子,被氧化生成O2。从能级图上看[108],H+/H2标准氢电极电势为0,O2/H2O标准氢电极电势为1.23 V。因此,选择的光催化剂要满足2个条件[6, 109]:(1)催化剂的禁带宽度要大于水的电解电压(1.23 V);(2)催化剂价带和导带的位置要分别与O2/H2O和H+/H2的电极电势相适宜。
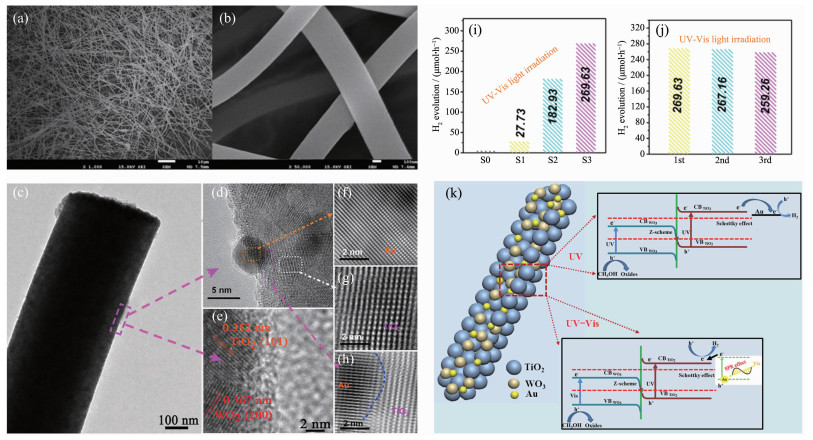
在光催化水分解的研究中,石墨烯基光催化剂[110-111]、金属磷化物[4, 112]、g-C3N4基光催化剂[113]、金属氧化物[114]、TiO2基光催化剂[115]等都是现在的热点材料。在众多催化剂中,TiO2作为最早的水分解的光催化剂,展现出较高的制氢效率。目前商业化的TiO2(P25)在制氢效率上处于许多材料之上,但可见光利用率低的问题依然限制了TiO2在制氢中的应用。2011年,Science报道了Chen等[116]制备出黑色氢化的TiO2,他们在TiO2表面构造了一种无序化结构,提高了TiO2对太阳光的吸收,并在光催化制氢中表现出高的催化活性和稳定性,制氢速率为10 000 μmol·g-1·h-1,高于大部分光催化剂。Su等[117]制备的TiO2/C复合催化制氢效率达到11 440 μmol·g-1·h-1。TiO2基Z型异质结在继承TiO2的高催化活性的基础上,同时又可以引入窄带隙的半导体提高TiO2对可见光的吸收,并进一步地提高TiO2的光催化活性。譬如,Zhang等[95]采用了静电纺丝的方法,制备了TiO2纳米纤维(S0)和TiO2/WO3复合纳米纤维(S1),并在TiO2/WO3复合纳米纤维修饰上Au的纳米颗粒,通过调控前驱体HAuCl4的质量分数(0.3%(S2)和0.45%(S3)),得到Au含量比例不同的TiO2/WO3/Au纳米纤维。图 13(a~c)呈现出S2的纳米纤维结构,通过HRTEM(图 13(d~g))可以明显的观察到TiO2的(101)晶面、WO3的(200)晶面以及Au的晶格条纹。图 13h也说明了Au与TiO2之间存在良好的接触,这为电子在两者间的转移创造了良好的前提条件。在紫外和可见光的照射下,测试光催化剂的制氢效率(图 13i)和稳定性(图 13j)。结果显示,TiO2/WO3相对于TiO2制氢速率提高到27.73 μmol·h-1,再引入Au后,催化剂的制氢速率提高到182.93 μmol·h-1。在稳定性上,S3样品3次循环的制氢速率一致。该催化剂具有较高的制氢活性的原因包含以下4个:(1)催化剂的结构为间接Z型异质结(图 13k),减小了电子与空穴的复合几率;(2) Au纳米颗粒所具有的等离子共振效应,增加了催化剂对可见光的吸收;(3) Au作为电子媒介,接受来自TiO2上的电子;(4)甲醇等空穴捕获剂的加入,进一步抑制了电子与空穴的复合。除了TiO2/WO3/Au光催化剂,许多TiO2基Z型异质结也表现出优异的制氢活性。其中含有0.5%(w/w)的Cu、Zn掺杂的CuZn-TiO2[96]在自然光的照射下,制氢速率达到了14 521 μmol·g-1·h-1。WO3-x量子点/TiO2[94]在300 W氙灯照射下,也表现出高达17 700 μmol·g-1·h-1的制氢速率。
构建TiO2基Z型异质结有利于解决TiO2可见光利用率低和电子-空穴易复合等缺点。另外,研究者在进行水分解的实验中,会加入空穴捕获剂,其目的是让空穴被捕获剂消耗掉,间接地减小电子与空穴的复合几率。常用的空穴捕获剂有硫化钠(Na2S)、亚硫酸钠(Na2SO3)[118]、三乙醇胺(TEOA)[119]、甲醇和乳酸[120]等。同时,也加入贵金属Pt[107, 121, 122]作为助催化剂,加速电子向外部迁移,有利于氢气的产生。
CO2是大气的主要成分,但化石燃料燃烧排放的大量CO2是引起温室效应的主要原因。1979年,Inoue等[123]首次采用光电技术将CO2还原为有机化合物,如甲酸、甲醛、甲醇和甲烷。这一方案的提出既解决了CO2的过量排放问题,又使还原生成的有机化合物作为新的能源被继续利用。近年来,随着太阳能资源的开发和利用,通过光催化实现CO2还原成为研究的热点领域之一。我们总结整理了相关的研究工作,提出了CO2还原存在以下7个还原反应[8, 124-125]:
|
${\rm{C}}{{\rm{O}}_2} + {{\rm{e}}^ - } \to \cdot {\rm{CO}}_2^ - \quad {E^ \ominus } = - 1.90{\rm{V}} $ |
(11) |
|
${\rm{C}}{{\rm{O}}_2} + 2{{\rm{H}}^ + } + 2{{\rm{e}}^ - } \to {\rm{CO}} + {{\rm{H}}_2}{\rm{O}}\quad {E^ \ominus } = - 0.53{\rm{V}} $ |
(12) |
|
${\rm{C}}{{\rm{O}}_2} + 2{{\rm{H}}^ + } + 2{{\rm{e}}^ - } \to {\rm{HC}}{{\rm{O}}_2}{\rm{H}}\quad {E^ \ominus } = - 0.61{\rm{V}} $ |
(13) |
|
${\rm{C}}{{\rm{O}}_2} + 4{{\rm{H}}^ + } + 4{{\rm{e}}^ - } \to {\rm{HCHO}} + {{\rm{H}}_2}{\rm{O}}\quad {E^ \ominus } = - 0.48{\rm{V}} $ |
(14) |
|
${\rm{C}}{{\rm{O}}_2} + 6{{\rm{H}}^ + } + 6{{\rm{e}}^ - } \to {\rm{C}}{{\rm{H}}_3}{\rm{OH}} + {{\rm{H}}_2}{\rm{O}}\quad {E^ \ominus } = - 0.38{\rm{V}} $ |
(15) |
|
${\rm{C}}{{\rm{O}}_2} + 8{{\rm{H}}^ + } + 8{{\rm{e}}^ - } \to {\rm{C}}{{\rm{H}}_4} + 2{{\rm{H}}_2}{\rm{O}}\quad {E^ \ominus } = - 0.24{\rm{V}} $ |
(16) |
|
$2{{\rm{H}}^ + } + 2{{\rm{e}}^ - } \to {{\rm{H}}_2}\quad {E^ \ominus } = - 0.42{\rm{V}} $ |
(17) |
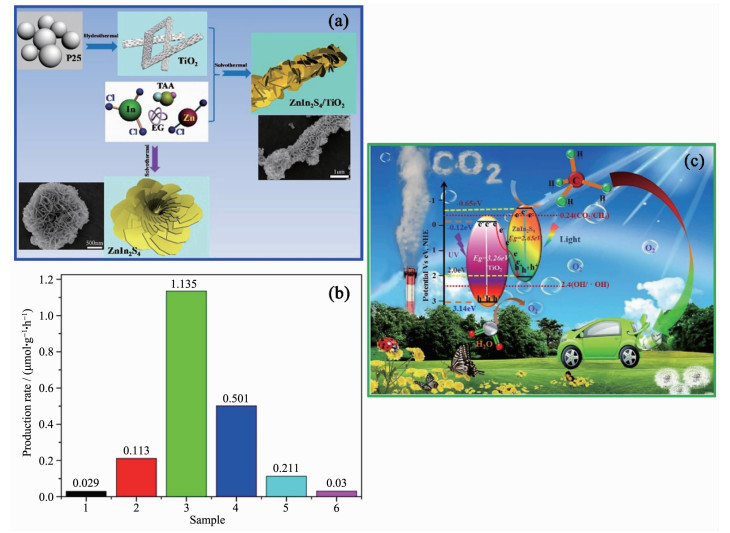
CO2单电子捕获的标准氢电极电势($E^{\ominus}$)为-1.90 V(pH=7.0),一般光催化剂的导带所处的能级均低于-1.90 V,导带上的电子很难被CO2所捕获,因此单电子还原过程很难实现,需要寻求其它反应途径。反应(12~16)为多电子还原,反应的$E^{\ominus}$在-0.5 V左右,反应相对容易发生。而反应发生的先后顺序与$E^{\ominus}$有关,电势越负,反应越难进行,因此多电子还原反应中,CO2的还原产物出现的先后顺序为CH4、CH3OH、CO和HCO2H。另外,多电子反应的$E^{\ominus}$都比较接近,因此各反应间存在竞争关系,而在水体系下的CO2还原,也需要考虑制氢反应(17)与其它反应的竞争,最终各反应的相互竞争导致CO2还原的选择性不高,但CO2还原的潜在应用价值依然吸引着研究者们不断地去寻找合适的光催化剂并应用在CO2还原上。随着光催化CO2还原的持续发展,相继报道出许多光催化剂,金属有机框架(MOF)光催化剂[125]、金属复合光催化剂[123]、g-C3N4基光催化剂[126]、石墨烯基光催化剂[127]和TiO2基光催化剂[41, 65, 98-99, 124]等,TiO2基Z型异质结作为其中一种光催化剂对CO2还原有较高的催化活性。Yang等[99]通过水热法将超薄的二维ZnIn2S4纳米片,复合在一维的TiO2纳米带上,形成了一种三维的Z型光催化剂ZnIn2S4/TiO2(图 14a)。ZnIn2S4的能带间隙为2.65 eV,它的截止吸收波长为468 nm,因此在Z型异质结中作为可见光的响应物质。根据ZnIn2S4和TiO2的能带分布(图 14c),TiO2导带上的电子与ZnIn2S4价带上的空穴复合,最终保留着TiO2价带上的空穴和ZnIn2S4导带上的电子。ZnIn2S4导带上的电子所处的电势为-0.65 eV,低于CO2多电子还原的电势。因而,从热力学角度而言,反应(12~16)的是可能发生的。在考察光催化剂还原CO2生成CH4的速率的实验中(图 14b),与单一的材料近乎为零的生成速率相比较而言,Z型异质结的CH4生成速率有明显的提高。同时,ZnIn2S4与TiO2的比例也是影响CH4的生成速率的关键因素,当ZnIn2S4与TiO2的物质的量之比为1:3时,CH4的生成速率达到最高,为1.135 μmol·g-1·h-1。
TiO2基Z型异质结在不降低TiO2氧化还原能力的基础上,通过引入窄带隙的半导体,增强对可见光的吸收,以及通过调控异质结的能带结构去调节光生电子的能级位置,有选择地进行CO2的还原反应。当前,TiO2基Z型异质结在光催化CO2还原上的应用研究较少,但其依然在光催化CO2还原上具有很大的应用潜力。产物选择性低、光催化剂的稳定性差和光能转换率低等是阻碍其实际应用的主要因素,寻求突破这些阻碍的方法成为现阶段研究的主要目的。
光催化是一种将光能转换为化学能的绿色安全的技术,在解决环境污染和能源危机这两大全球性问题上具有广阔的前景。光催化剂TiO2基Z型异质结作为新型催化剂在继承TiO2的优势基础上,还具有氧化还原能力强、电子-空穴易分离和可见光利用率高(在引入窄带隙的半导体时)等性质,并在光催化降解污染物、水分解和CO2还原上得到了广泛的应用。
本文综述了TiO2光催化剂、TiO2异质结光催化剂、TiO2基Z型异质结光催化剂的能带排列与电荷转移规则,比较了type-Ⅱ异质结和Z型异质结的异同点,并在此基础上介绍了3种区分彼此的方法,最后归纳整理了部分TiO2基Z型异质结在光催化降解污染物、水分解和CO2还原上的应用。综合以上讨论,TiO2基Z型异质结具有许多独特的优势,但依然存在一些问题:(1)对探索Z型异质结的光催化机理,依然缺乏有效的手段和准确的理论支撑;(2) Z型异质结分离电子和空穴的前提是电子与空穴的复合,这样会导致催化剂表面载流子的浓度下降,从而降低催化活性;(3)如何有目的地构建TiO2基Z型异质结而不是type-Ⅱ异质结,依然是一个难题。
Fujishima A, Honda K. Nature, 1972, 238:37-38 doi: 10.1038/238037a0
Herrmann J M. Catal. Today, 1999, 53(1):115-129 https://www.researchgate.net/publication/222283180_Heterogeneous_photocatalysis_Fundamentals_and_applications_to_the_removal_of_various_types_of_aqueous_pollutants
Chong M N, Jin B, Chow C W K, et al. Water Res., 2010, 44(10):2997-3027 doi: 10.1016/j.watres.2010.02.039
Shi Y M, Zhang B. Chem. Soc. Rev., 2016, 45(6):1529-1541 doi: 10.1039/C5CS00434A
Zhu M S, Sun Z C, Fujitsuka M, et al. Angew. Chem. Int. Ed., 2018, 57(8):2160-2164 doi: 10.1002/anie.201711357
Fang M, Dong G F, Wei R J, et al. Adv. Energy Mater., 2017, 7(23):1700559 doi: 10.1002/aenm.201700559
Chen Y, Jia G, Hu Y F, et al. Sustainable Energy Fuels, 2017, 1(9):1875-1898 doi: 10.1039/C7SE00344G
Oh Y, Hu X L. Chem. Soc. Rev., 2013, 42(6):2253-2261 doi: 10.1039/C2CS35276A
Qi K Z, Cheng B, Yu J G, et al. Chinese J. Catal., 2017, 38(12):1936-1955 doi: 10.1016/S1872-2067(17)62962-0
Schneider J, Matsuoka M, Takeuchi M, et al. Chem. Rev., 2014, 114(19):9919-9986 doi: 10.1021/cr5001892
Cong Y, Li X K, Qin Y, et al. Appl. Catal. B, 2011, 107(1):128-134
Ohno T, Akiyoshi M, Umebayashi T, et al. Appl. Catal. A, 2004, 265(1):115-121
Cong Y, Zhang J L, Chen F, et al. J. Phys. Chem. C, 2007, 111(19):6976-6982 doi: 10.1021/jp0685030
Hong X T, Wang Z P, Cai W M, et al. Chem. Mater., 2005, 17(6):1548-1552 doi: 10.1021/cm047891k
Xu S C, Zhang Y X, Pan S S, et al. J. Hazard. Mater., 2011, 196:29-35 doi: 10.1016/j.jhazmat.2011.08.068
Dong J Y, Han J, Liu Y S, et al. ACS Appl. Mater. Interfaces, 2014, 6(3):1385-1388 doi: 10.1021/am405549p
Wu M C, Chih J S, Huang W K. CrystEngComm, 2014, 16(46):10692-10699 doi: 10.1039/C4CE01348D
Xing M Y, Yang B X, Yu H, et al. J. Phys. Chem. Lett., 2013, 4(22):3910-3917 doi: 10.1021/jz4021102
Yu J G, Qi L F, Jaroniec M. J. Phys. Chem. C, 2010, 114(30):13118-13125 doi: 10.1021/jp104488b
Cozzoli P D, Fanizza E, Comparelli R, et al. J. Phys. Chem. B, 2004, 108(28):9623-9630 doi: 10.1021/jp0379751
Ortel E, Sokolov S, Zielke C, et al. Chem. Mater., 2012, 24(20):3828-3838 doi: 10.1021/cm301081w
Yan B L, Zhang L J, Tang Z Y, et al. Appl. Catal. B, 2017, 218:743-750 doi: 10.1016/j.apcatb.2017.07.020
Wang M G, Han J, Hu Y M, et al. ACS Appl. Mater. Interfaces, 2016, 8(43):29511-29521 doi: 10.1021/acsami.6b10480
Khanchandani S, Kumar S, Ganguli A K. ACS Sustainable Chem. Eng., 2016, 4(3):1487-1499 doi: 10.1021/acssuschemeng.5b01460
Xu F Y, Zhang L Y, Cheng B, et al. ACS Sustainable Chem. Eng., 2018, 6(9):12291-12298 doi: 10.1021/acssuschemeng.8b02710
Wang C C, Zhan Y, Wang Z Y. Chemistry Select, 2018, 3(6):1713-1718 https://www.researchgate.net/publication/323095915_TiO_2_MoS_2_and_TiO_2_MoS_2_Heterostructures_for_Use_in_Organic_Dyes_Degradation
Lu D, Zhang G, K Wan Z. Appl. Surf. Sci., 2015, 358:223-230 doi: 10.1016/j.apsusc.2015.08.240
Cai J B, Wu X Q, Li S X, et al. ACS Sustainable Chem. Eng., 2016, 4(3):1581-1590 doi: 10.1021/acssuschemeng.5b01511
Mazierski P, Malankowska A, Kobylanski M, et al. ACS Catal., 2017, 7(4):2753-2764 doi: 10.1021/acscatal.7b00056
Zhang Y C, Li J, Xu H Y. Appl. Catal. B, 2012, 123-124:18-26 doi: 10.1016/j.apcatb.2012.04.018
Vinodgopal K, Kamat P V. Environ. Sci. Technol., 1995, 29(3):841-845 doi: 10.1021/es00003a037
Ong W J, Tan L L, Ng Y H, et al. Chem. Rev., 2016, 116(12):7159-7329 doi: 10.1021/acs.chemrev.6b00075
Low J X, Yu J G, Jaroniec M, et al. Adv. Mater., 2017, 29(20):1601694 doi: 10.1002/adma.201601694
Williams G, Seger B, Kamat P V. ACS Nano, 2008, 2(7):1487-1491 doi: 10.1021/nn800251f
Zhang Y H, Tang Z R, Fu X Z, et al. ACS Nano, 2010, 4(12):7303-7314 doi: 10.1021/nn1024219
Liang Y Y, Wang H L, Casalongue H S, et al. Nano Res., 2010, 3(10):701-705 doi: 10.1007/s12274-010-0033-5
Dai G P, Yu J G, Liu G. J. Phys. Chem. C, 2011, 115(15):7339-7346 doi: 10.1021/jp200788n
Zhang Y, Xie Y H, Li J, et al. J. Sol-Gel Sci. Technol., 2014, 71(1):38-42 doi: 10.1007/s10971-014-3328-2
Xing X L, Zhu H H, Zhang M, et al. Catal. Sci. Technol., 2018, 8(14):3629-3637 doi: 10.1039/C8CY01035H
Tan B Y, Ye X Z, Li Y J, et al. Chem. Eur. J., 2018, 24:1-12 doi: 10.1002/chem.201705257
Aguirre M E, Zhou R X, Eugene A J, et al. Appl. Catal. B, 2017, 217:485-493 doi: 10.1016/j.apcatb.2017.05.058
Hao J G, Zhang S F, Ren F, et al. J. Colloid Interface. Sci., 2017, 508:419-425 doi: 10.1016/j.jcis.2017.08.065
Liu J J, Cheng B, Yu J G. Phys. Chem. Chem. Phys., 2016, 18(45):31175-31183 doi: 10.1039/C6CP06147H
Reli M, Huo P W, Sihor M, et al. J. Phys. Chem. A, 2016, 120(43):8564-8573 doi: 10.1021/acs.jpca.6b07236
Lu L Y, Wang G H, Zou M, et al. Appl. Surf. Sci., 2018, 441:1012-1023 doi: 10.1016/j.apsusc.2018.02.080
Zhang G H, Zhang T Y, Li B, et al. Appl. Surf. Sci., 2018, 433:963-974 doi: 10.1016/j.apsusc.2017.10.135
Wei H, McMaster W A, Tan J Z Y, et al. J. Phys. Chem. C, 2017, 121(40):22114-22122 doi: 10.1021/acs.jpcc.7b06493
Jiang G D, Yang X X, Wu Y, et al. Mol. Catal., 2017, 432:232-241 doi: 10.1016/j.mcat.2016.12.026
Tang Q, Meng X F, Wang Z Y, et al. Appl. Surf. Sci., 2018, 430:253-262 doi: 10.1016/j.apsusc.2017.07.288
Wang C L, Hu L M, Chai B, et al. Appl. Surf. Sci., 2018, 430:243-252 doi: 10.1016/j.apsusc.2017.08.036
Gu W L, Lu F X, Wang C, et al. ACS Appl. Mater. Interfaces, 2017, 9(34):28674-28684 doi: 10.1021/acsami.7b10010
Ma L N, Wang G H, Jiang C J, et al. Appl. Surf. Sci., 2018, 430:263-272 doi: 10.1016/j.apsusc.2017.07.282
Troppova I, Sihor M, Reli M, et al. Appl. Surf. Sci., 2018, 430:335-347 doi: 10.1016/j.apsusc.2017.06.299
Wang W, Fang J J, Shao S F, et al. Appl. Catal. B, 2017, 217:57-64 doi: 10.1016/j.apcatb.2017.05.037
Yan J Q, Wu H, Chen H, et al. Appl. Catal. B, 2016, 191:130-137 doi: 10.1016/j.apcatb.2016.03.026
Li J, Zhang M, Li Q Y, et al. Appl. Surf. Sci., 2017, 391:184-193 doi: 10.1016/j.apsusc.2016.06.145
Yu J G, Wang S H, Low J X, et al. Phys. Chem. Chem. Phys., 2013, 15(39):16883-16890 doi: 10.1039/c3cp53131g
Zheng X T, Yang L M, Li Y B, et al. Electrochim. Acta, 2019, 298:663-669 doi: 10.1016/j.electacta.2018.12.130
Li J, Zhang M, Li X, et al. Appl. Catal. B, 2017, 212:106-114 doi: 10.1016/j.apcatb.2017.04.061
Wang J, Wang G H, Wei X H, et al. Appl. Surf. Sci., 2018, 456:666-675 doi: 10.1016/j.apsusc.2018.06.182
Li Y H, Lv K L, Ho W K, et al. Appl. Catal. B, 2017, 202:611-619 doi: 10.1016/j.apcatb.2016.09.055
Li Q, Xia Y, Yang C, et al. Chem. Eng. J., 2018, 349:287-296 doi: 10.1016/j.cej.2018.05.094
Meng S G, Sun W T, Zhang S J, et al. J. Phys. Chem. C, 2018, 122(46):26326-26336 doi: 10.1021/acs.jpcc.8b07524
Zhang Z, Yates J T. Chem. Rev., 2012, 112(10):5520-5551 doi: 10.1021/cr3000626
Xu F Y, Zhang J J, Zhu B C, et al. Appl. Catal. B, 2018, 230:194-202 doi: 10.1016/j.apcatb.2018.02.042
Tersoff J. Phys. Rev. B:Condens. Matter, 1985, 32(10):6968-6971 doi: 10.1103/PhysRevB.32.6968
Low J X, Dai B Z, Tong T, et al. Adv. Mater., 2019, 31(6):1802981 doi: 10.1002/adma.201802981
Jiang W S, Zong X P, An L, et al. ACS Catal., 2018, 8(3):2209-2217 doi: 10.1021/acscatal.7b04323
Meng A Y, Zhu B C, Zhong B, et al. Appl. Surf. Sci., 2017, 422:518-527 doi: 10.1016/j.apsusc.2017.06.028
Hu Z Z, Quan H H, Chen Z, et al. Photochem. Photobiol. Sci., 2018, 17(1):51-59 doi: 10.1039/C7PP00256D
Qin N, Liu Y H, Wu W M, et al. Langmuir, 2015, 31(3):1203-1209 doi: 10.1021/la503731y
Zhang J, Zhou D D, Dong S S, et al. J. Hazard. Mater., 2019, 366:311-320 doi: 10.1016/j.jhazmat.2018.12.013
Han X G, Kuang Q, Jin M S, et al. J. Am. Chem. Soc., 2009, 131(9):3152-3153 doi: 10.1021/ja8092373
Duan Y Y, Liang L, Lv K L, et al. Appl. Surf. Sci., 2018, 456:817-826 doi: 10.1016/j.apsusc.2018.06.128
Bard A J. J. Photochem., 1979, 10(1):59-75 https://www.researchgate.net/publication/222784892_Photoelectrochemistry_and_heterogeneous_photo-catalysis_at_semiconductors?ev=auth_pub
Xu Q L, Zhang L Y, Yu J G, et al. Mater. Today, 2018, 21(10):1042-1063 doi: 10.1016/j.mattod.2018.04.008
Tada H, Mitsui T, Kiyonaga T, et al. Nat. Mater., 2006, 5:782-786 doi: 10.1038/nmat1734
Wu F J, Li X, Liu W, et al. Appl. Surf. Sci., 2017, 405:60-70 doi: 10.1016/j.apsusc.2017.01.285
Liu X, Wang Z Q, Wu Y Z, et al. J. Colloid Interface Sci., 2019, 538:689-698 doi: 10.1016/j.jcis.2018.12.070
Zou Y J, Shi J W, Ma D D, et al. ChemCatChem, 2017, 9(19):3752-3761 doi: 10.1002/cctc.201700542
Zhao W, Liu J C, Deng Z Y, et al. Int. J. Hydrogen Energy, 2018, 43(39):18232-18241 doi: 10.1016/j.ijhydene.2018.08.026
Gao H Q, Zhang P, Hu J H, et al. Appl. Surf. Sci., 2017, 391:211-217 doi: 10.1016/j.apsusc.2016.06.170
Guo Y R, Xiao L M, Zhang M, et al. Appl. Surf. Sci., 2018, 440:432-439 doi: 10.1016/j.apsusc.2018.01.144
Bai S, Wang L L, Li Z Q, et al. Adv. Sci., 2017, 4(1):1600216 doi: 10.1002/advs.201600216
Kong L N, Zhang X T, Wang C H, et al. Appl. Surf. Sci., 2018, 448:288-296 doi: 10.1016/j.apsusc.2018.04.011
Wang G, Zhang L J, Yan B L, et al. ChemCatChem, 2019, 11(3):1057-1063
Li J Z, Zhong J B, Si Y J, et al. Solid State Sci., 2016, 52:106-111 doi: 10.1016/j.solidstatesciences.2015.12.020
Liao W J, Murugananthan M, Zhang Y R. Phys. Chem. Chem. Phys., 2015, 17(14):8877-8884 doi: 10.1039/C5CP00639B
Chen B H, Li P R, Zhang S S, et al. J. Colloid Interface Sci., 2016, 478:263-270 doi: 10.1016/j.jcis.2016.05.053
Chi C Y, Pan J Q, You M Z, et al. J. Phys. Chem. Solids, 2018, 114:173-178 doi: 10.1016/j.jpcs.2017.11.028
Wang F L, Wu Y L, Wang Y F, et al. Chem. Eng. J., 2019, 356:857-868 doi: 10.1016/j.cej.2018.09.092
Hu J H, Wang L J, Zhang P, et al. J. Power Sources, 2016, 328:28-36 doi: 10.1016/j.jpowsour.2016.08.001
Xu F Y, Xiao W, Cheng B, et al. Int. J. Hydrogen Energy, 2014, 39(28):15394-15402 doi: 10.1016/j.ijhydene.2014.07.166
Pan L, Zhang J W, Jia X, et al. Chin. J. Catal., 2017, 38(2):253-259
Gao H Q, Zhang P, Zhao J T, et al. Appl. Catal. B, 2017, 210:297-305 doi: 10.1016/j.apcatb.2017.03.050
Subha N, Mahalakshmi M, Myilsamy M, et al. Appl. Catal. A, 2018, 553:43-51 doi: 10.1016/j.apcata.2018.01.009
Zhao J T, Zhang P, Wang Z, et al. Sci. Rep., 2017, 7:16116 doi: 10.1038/s41598-017-12203-y
Wang D, Pan X Y, Wang G T, et al. RSC Adv., 2015, 5(28):22038-22043 doi: 10.1039/C4RA15215H
Yang G, Chen D M, Ding H, et al. Appl. Catal. B, 2017, 219:611-618 doi: 10.1016/j.apcatb.2017.08.016
Konstantinou I K, Albanis T A. Appl. Catal. B, 2004, 49(1):1-14 https://www.researchgate.net/publication/222957082_TiO2-Assisted_Photocatalytic_Degradation_of_Azo_Dyes_in_Aqueous_Solution_Kinetic_and_Mechanistic_Investigations_A_Review
Chen C C, Ma W H, Zhao J C. Chem. Soc. Rev., 2010, 39(11):4206-4219 doi: 10.1039/b921692h
崔言娟, 王愉雄, 王浩, 等.化学进展, 2016, 4:428-437 doi: 10.7536/PC151025CUI Yan-Juan, WANG Yu-Xiong, WANG Hao, et al. Prog. Chem., 2016, 4:428-437 doi: 10.7536/PC151025
Yan L, Gu Z J, Zheng X P, et al. ACS Catal., 2017, 7(10):7043-7050 doi: 10.1021/acscatal.7b02170
Lin H, Huang C P, Li W, et al. Appl. Catal. B, 2006, 68(1/2):1-11 https://www.sciencedirect.com/science/article/pii/S0926337306003328
张晓东, 杨阳, 李红欣, 等.化学进展, 2016, 10:1550-1559ZHANG Xiao-Dong, YANG Yang, LI Hong-Xin, et al. Prog. Chem., 2016, 10:1550-1559
Man X L, Wu R L, Lv H H, et al. J. Appl. Polym. Sci., 2015, 132(41):42627 https://www.researchgate.net/publication/280916236_Synthesis_of_a_montmorillonite-supported_titania_nanocomposite_with_grafted_cellulose_as_a_template_and_its_application_in_photocatalytic_degradation?ev=auth_pub
Martin D J, Qiu K P, Shevlin S A, et al. Angew. Chem. Int. Ed., 2014, 53(35):9240-9245 doi: 10.1002/anie.201403375
Hisatomi T, Kubota J, Domen K. Chem. Soc. Rev., 2014, 43(22):7520-7535 doi: 10.1039/C3CS60378D
Yang Y, Niu S W, Han D D, et al. Adv. Energy Mater., 2017, 7(19):1700555 doi: 10.1002/aenm.201700555
Lu K Q, Xin X, Zhang N, et al. J. Mater. Chem. A, 2018, 6(11):4590-4604 doi: 10.1039/C8TA00728D
Li X, Shen R C, Ma S, et al. Appl. Surf. Sci., 2018, 430:53-107 doi: 10.1016/j.apsusc.2017.08.194
Cao S, Wang C J, Fu W F, et al. ChemSusChem, 2017, 10(22):4306-4323 doi: 10.1002/cssc.201701450
Naseri A, Samadi M, Pourjavadi A, et al. J. Mater. Chem. A, 2017, 5(45):23406-23433 doi: 10.1039/C7TA05131J
Wang F Y, Song L F, Zhang H C, et al. J. Electron. Mater., 2017, 46(8):4716-4724 doi: 10.1007/s11664-017-5491-z
Singh R, Dutta S. Fuel, 2018, 220:607-620 doi: 10.1016/j.fuel.2018.02.068
Chen X B, Liu L, Yu P Y, et al. Science, 2011, 331(6018):746-750 doi: 10.1126/science.1200448
Yan B L, Zhou J, Liang X Y, et al. Appl. Surf. Sci., 2017, 392:889-896 doi: 10.1016/j.apsusc.2016.09.117
Wei R B, Huang Z L, Gu G H, et al. Appl. Catal. B, 2018, 231:101-107 doi: 10.1016/j.apcatb.2018.03.014
Zhang X Q, Luo D, Zhang W Y, et al. Appl. Catal. B, 2018, 232:371-383 doi: 10.1016/j.apcatb.2018.03.070
Chen W X, Fang J S, Zhang Y W, et al. Nanoscale, 2018, 10(9):4463-4474 doi: 10.1039/C7NR08943K
Liu G, Niu P, Sun C H, et al. J. Am. Chem. Soc., 2010, 132(33):11642-11648 doi: 10.1021/ja103798k
Yang P J, Zhao J H, Qiao W, et al. Nanoscale, 2015, 7(45):18887-18890 doi: 10.1039/C5NR05570A
Takeda H, Cometto C, Ishitani O, et al. ACS Catal., 2017, 7(1):70-88 https://www.researchgate.net/publication/309541063_Electrons_Photons_Protons_and_Earth_Abundant_Metal_Complexes_for_Molecular_Catalysis_of_CO_2_Reduction
Sohn Y K, Huang W X, Taghipour F. Appl. Surf. Sci., 2017, 396:1696-1711 doi: 10.1016/j.apsusc.2016.11.240
Crake A. Mater. Sci. Technol., 2017, 33(15):1737-1749 doi: 10.1080/02670836.2017.1318250
Sun Z X, Wang H Q, Wu Z B A, et al. Catal. Today, 2018, 300:160-172 doi: 10.1016/j.cattod.2017.05.033
Low J X, Yu J G, Ho W K. J. Phys. Chem. Lett., 2015, 6(21):4244-4251 doi: 10.1021/acs.jpclett.5b01610

图 2 TiO2光催化中的光诱导反应及其时间尺度[10]
Figure 2 Photoinduced reactions in TiO2 photocatalysis and the corresponding time scales[10]
ehot- and hhot+ are excited electrons and holes, respectively; eCB- and hCB+ are electrons in conduction band and holes in valence band, respectively; eshallow-, hshallow+, edeep- and hdeep+: are electrons and holes in shallow or deep trap, respectively; etr- and htr+ are trapped electrons and holes, including etrshallow-, htrshallow+, etrdeep- and htrdeep+; rec is short for recombination

图 3 异质结的3种类型
Figure 3 Three types of heterojunctions in a typical semiconductor hybrid nanocomposite

图 4 (a) type-Ⅱ异质结和(b) Z型异质结的能带排列和电子迁移机理
Figure 4 Band arrangement and electron migration mechanism of (a) type-Ⅱ heterojunctions and (b) Z-scheme heterojunctions

图 5 Cu2O/TiO2复合物分别为(a) type-Ⅱ异质结和(b) Z型异质结的光催化机理; (c) Cu2O (上)、纯TiO2 (中)、Cu2O/TiO2异质结构(下)的紫外光电子能谱,左右两侧的外推分别对应价带和次级电子谱的起始值; (d)通过7-羟基香豆素的荧光光谱探测经λ≥305 nm光照射后Cu2O/TiO2表面·OH的存在以及相对应的·OH产量[41]
Figure 5 (a) Type-Ⅱ mechanism and (b) direct Z-scheme mechanism of Cu2O/TiO2 composites; (c) Ultraviolet photoelectron spectra for Cu2O octahedral (top), pure TiO2 (center), and Cu2O/TiO2 heterostructure (bottom). Extrapolations to the left and right hand sides correspond to the onset values for the valence band and secondary electron spectra, respectively; (d) Fluorescence spectra of 7-hydroxycoumarin for probing ·OH on the surface Cu2O/TiO2 during irradiation at λ≥305 nm and the corresponding production amount of ·OH[41]

图 6 Z型异质结TiO2/g-C3N4纳米棒阵列的SEM图(a)和光催化机理图(c)[42]; type-Ⅱ异质结TiO2/g-C3N4的TEM图(b)和光催化机理图(d)[44]
Figure 6 SEM image (a) and photocatalytic mechanism diagram (c) of Z-scheme heterojunction TiO2/g-C3N4 nanorods arrays[42]; TEM image (b) and photocatalytic mechanism (d) of type-Ⅱ heterojunction TiO2/g-C3N4[44]

图 7 (a) type-Ⅱ异质结中电荷转移和分离的过程; (b)提出了直接Z-scheme异质结MoSe2@TiO2纳米管阵列(TNTs)复合物在人造太阳光照射下光催化降解4-NP的机理[58]
Figure 7 (a) Scheme illustration for the charge transfer and separation in conventional type-Ⅱ heterojunction; (b) Proposed mechanism for the photocatalytic degradation of 4-NP over MoSe2@TiO2 nanotube arrays (TNTs) direct Z-scheme composite under artificial solar light irradiation[58]

图 8 计算(a)TiO2 (101)面和(b) CuInS2 (004)面的静电势; (c)在模拟太阳光照射下, TiO2/CulnS2复合物中电荷转移和分离的示意图[65]
Figure 8 Calculated electrostatic potentials for (a) TiO2 (101) face and (b) CuInS2 (004) face; (c) Schematic illustration of the charge transfer and separation in TiO2/CulnS2 composites under simulated sunlight light irradiation[65]

图 9 (a) CdS、TiO2、TiO2/CdS、P25样品的光催化性能比较; (b, c) TiO2/CdS在黑暗或365 nm LED灯照射下(UV-TiO2/CdS)的Ti2p (b)和Cd3d (c)的高分辨率XPS谱图; (d)光照下,以CdS、TiO2、TiO2/CdS为光催化剂,7-羟基香豆素在457 nm处光致发光强度随时间的变化; TiO2/CdS复合材料作为type-Ⅱ (e)和直接Z-scheme (f)异质结的电荷载流子迁移机理示意图[67]
Figure 9 (a) Comparison of the photocatalytic performance of the CdS, TiO2, TiO2/CdS, and P25 samples; (b, c) High-resolution XPS for Ti2p (b) and Cd3d (c) of TiO2/CdS in the dark or under 365 nm LED irradiation (UV-TiO2/CdS); (d) Changes of the 7-hydroxycoumarin photoluminescence intensity at 457 nm using CdS, TiO2, and TiO2/CdS as photocatalysts under light irradiation over time; Schematic illustration of the charge-carrier migration mechanism according to the type-Ⅱ (e) and direct Z-scheme (f) heterojunction for the TiO2/CdS composite[67]


图 12 TiO2@g-C3N4核壳纳米棒阵列的(a)合成过程, (b)在可见光下各催化剂对RhB的光催化降解曲线和(c)作为Z型光催化剂的降解机理图[42]; (d)纳米纤维状的TiO2/g-C3N4异质结光催化剂的制备流程, (e) TiO2组成比例不同的TiO2/g-C3N4样品在复合太阳光下对RhB的光催化降解曲线和(f) Z型异质结TiO2/g-C3N4光催化剂的光催化机理[50]
Figure 12 (a) Schematic illustration of synthesis process of TiO2@g-C3N4 core-shell nanorod arrays, (b) photocatalytic activity plots of TiO2 and TiO2@g-C3N4 nanorod arrays for degradation of RhB dye under visible light, and (c) degradation mechanism of g-C3N4@TiO2 in the form of Z-scheme[42]; (d) Schematic representation of the fabrication of electrospun nanofibrous TiO2/g-C3N4 heterojunction photocatalysts, (e) photocatalytic degradation RhB aqueous solution over different TiO2/g-C3N4 photocatalysts under simulated solar light irradiation, and (f) schematic showing the Z-scheme photocatalytic mechanism of the nanofibrous TiO2/g-C3N4 heterojunction photocatalys[50]

图 13 S2样品的(a, b)SEM图, (c)低倍TEM图以及(d~h) HRTEM图; 不同样品在紫外-可见光照射下的光催化制氢活性(i)以及稳定性(j); (k) Z型异质结TiO2/WO3/Au纳米纤维光催化制氢的机理图[95]
Figure 13 (a, b) SEM images, (c) a low-resolution TEM image and (d~h) HRTEM images of as-prepared S2 sample; photocatalytic hydrogen production activity (i) and stability (j) of different samples under UV-Vis lightirradiation; (k) schematic diagram of the photocatalytic H2 generation over the Z-scheme heterojunctionTiO2/WO3/Au composite nanofibers[95]

图 14 (a) 三维直接Z型光催化剂ZnIn2S4纳米片/TiO2纳米带的制备示意图; (b)甲烷的生成速率图; (1) ZnIn2S4纳米片, (2) ZIS-0.25/TO (ZnIn2S4/TiO2的物质的量比值nZnIn2S4/nTiO2=0.25), (3) ZIS-0.33/TO (nZnIn2S4/nTiO2=0.33), (4) ZIS-0.50/TO (nZnIn2S4/nTiO2=0.50), (5) ZIS-1/TO (nZnIn2S4/nTiO2=1)和(6) TiO2纳米带; (c)紫外和可见光照射下的ZnIn2S4纳米片/TiO2纳米带光催化机理图[99]
Figure 14 Schematic illustration (a) of the fabrication of 3D ZnIn2S4 nanosheets/TiO2 nanobelts direct Z-scheme photocatalysts; (b) CH4 generation velocity over (1) ZnIn2S4 nanosheets, (2) ZIS-0.25/TO (nZnIn2S4/nTiO2 (molar radio of ZnIn2S4 to TiO2)=0.25), (3) ZIS-0.33/TO (nZnIn2S4/nTiO2=0.33), (4) ZIS-0.50/TO (nZnIn2S4/nTiO2=0.50), (5) ZIS-1/TO (nZnIn2S4/nTiO2=1) and (6) TiO2 nanobelts; (c) Schematic diagrams for photocatalysis of ZnIn2S4 nanosheets/TiO2 nanobelts under UV-Vis irradiation[99]
表 1 不同TiO2/g-C3N4光催化剂的异质结类型和应用
Table 1. Types of heterojunction and applications of different TiO2/g-C3N4photocatalysts
| Photocatalyst | Type of heterojunction | Application | Reference |
| TiO2@g-C3N4 core-shell nanorods arrays | Z-scheme | Degradation of RhB | [42] |
| TiO2g-C3N4 | Type-Ⅱ | Reduction of CO2 and decomposition of N2O | [44] |
| Hierarchical TiO2/g-C3N4 hybrids | Z-scheme | Degradation of RhB | [45] |
| TiO2/g-C3N4 | Type-Ⅱ | Degradation of MO | [46] |
| Mesoporous TiO2/g-C3N4 microspheres | Type-Ⅱ | Degradation of phenol | [47] |
| Spherical TiO2/g-C3N4 | Type-Ⅱ | Decomposition of MB | [48] |
| TiO2/g-C3N4 nanofibers | Z-scheme | Degradation of RhB | [49] |
| Nanofibrous TiO2/g-C3N4 | Z-scheme | Degradation of RhB | [50] |
| Ultrathin g-C3N4/anatase TiO2 nanosheets | Type-Ⅱ | Degradation of MB | [51] |
| Core-shell TiO2@g-C3N4 hollow microspheres | Type-Ⅱ | Degradation of RhB | [52] |
| TiO2/g-C3N4 | Z-scheme | Decomposition of N2O | [53] |
| TiO2@g-C3N4 core-shell quantum heterojunction | Type-Ⅱ | Degradation of tetracycline | [54] |
| TiO2/C3N4 | Z-scheme | Water splitting | [55] |
| g-C3N4-TiO2 | Z-scheme | Remove of propylene | [56] |
| g-C3N4-TiO2 | Z-scheme | Decomposition of formaldehyde | [57] |
| RhB: rhodamine B; MB: methylene blue; MO: methyl orange | |||
 下载: 导出CSV
下载: 导出CSV
表 2 TiO2基Z型异质结在光催化中的应用
Table 2. Applications of TiO2 based Z-scheme heterojunctions in photocatalysis
| Photocatalyst | Light source | Application | Photocatalytic efficiency | Reference |
| TiO2@g-C3N4 core-shell nanorod arrays | 100 W Xe lamp | Degradation of RhB | 180 min, 95.68% | [42] |
| Hierarchical TiO2/g-C3N4 | 350 W Xe arc lamp | Degradation of RhB | 0.055 min-1 | [45] |
| TiO2/g-C3N4 nanofibers | 300 W Xe lamp | Degradation of RhB | 70 min, 99% | [49] |
| Nanofibrous TiO2/g-C3N4 | 500 W Xe lamp | Degradation of RhB | 0.053 45 min-1 | [50] |
| TiO2/g-C3N4 | 8 W Hg lamp | Photocatalytic N2O | 0.001 1 min-1 | [53] |
| BiOI/TiO2 | 500 W Xe lamp | Degradation of MO | 25 min, 85.0% | [87] |
| g-C3N4-Ti3+/TiO2 nanotube arrays | 11 W incandescent lamp | Degradation of phenol | 7 h, 74% | [88] |
| g-C3N4-RGO-TiO2 | 300 W Xe lamp | Degradation of MB | 0.013 7 min-1 | [78] |
| Rutile TiO2/g-C3N4 quantum dots | 500 W Xe lamp | Degradation of RhB | 0.011 5 min-1 | [61] |
| g-C3N4 nanosheets/TiO2 nanosheets | 300 W Xe lamp | Degradation of MO | 0.037 31 min-1 | [89] |
| TiO2 nanotubes/Ag3PO4 | Sunlight irradiation | Degradation of MB | 0.048 min-1 | [90] |
| Nitrogen-doped carbon dots/{001} TiO2 nanosheets |
350 W Xe lamp | Degradation of diclofenac | 60 min, 91.5% | [91] |
| Carbon-modified TiO2/WO3 nanofibers | 300 W Xe arc lamp | Hydrogen production | ~1 570 μmol·g-1·h-1 for H2 | [92] |
| TiO2/WO3/Pt | 300 W Xe arc lamp | Hydrogen production | 128.66 μmol·g-1·h-1 | [82] |
| Anatase/rutile TiO2 nanofibers | 350 W Xe arc lamp | Hydrogen production | 6 480 μmol·g-1·h-1 | [93] |
| WO3-x quantum dots/TiO2 | 300 W Xe lamp | Hydrogen production | 17 700 μmol·g-1·h-1 | [94] |
| TiO2/WO3/Au nanofibers | 300 W Xe arc lamp | Hydrogen production | 5 393 μmol·g-1·h-1 | [95] |
| CuZn-TiO2 | solar light | Hydrogen production | 14 521 μmol·g-1·h-1 | [96] |
| Au/Pt/WO3/TiO2 nanofibers | 300 W Xe arc lamp | Hydrogen production | 242.09 μmol·g-1·h-1 | [97] |
| g-C3N4/TiO2 | 300 W Xe lamp | Hydrogen production | 12 600 μmol·g-1·h-1 for H2 | [59] |
| TiO2/C3N4 | 150 W Xe lamp | Water splitting | 251 μmol·g-1·h-1 for H2 | [55] |
| 121.5 μmol·g-1·h-1 for O2 | ||||
| TiO2/Cu2O | 1 kW Hg (Xe) arc lamp | Reduction of CO2 | 2.11 μmol·g-1·h-1 for CO | [41] |
| Cu2O/TiO2 | 300 W Xe lamp | Photocatalytic propane | 0.54 min-1 for C3H6 | [98] |
| TiO2/CuInS2 | 350 W Xe arc lamp | Reduction of CO2 | 2.5 μmol·g-1·h-1 for CH | [65] |
| 0.86 μmol·g-1·h-1 for CH3OH | ||||
| ZnIn2S4/TiO2 | 300 W Xe lamp | Reduction of CO2 | 1.135 μmol·g-1·h-1 for CH4 | [99] |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们