Table 1.
Crystallographic data and refinement parameters for complexes 1~3 and 6
Citation:
LI Song, GAN Xian-Xue, TANG Liang-Fu. Syntheses and Catalytic Properties of Metal Carbonyl Derivatives with Hydroxymethyl Functionalized Pyrazoles[J]. Chinese Journal of Inorganic Chemistry,
2018, 34(8): 1573-1580.
doi:
10.11862/CJIC.2018.183

羟甲基功能化吡唑金属羰基化合物的合成及催化性质
摘要:
研究了3(5)-羟甲基-5(3)-甲基吡唑(L1),4-羟甲基吡唑(L2)及双(3-羟甲基-5-甲基吡唑)甲烷(L3)与羰基钨(钼)的反应,合成了一系列以单齿及双齿氮配位的羰基金属衍生物LW(CO)5(L=L1或L2)和L3M(CO)4(M=Mo或W)。通过核磁、红外及X射线单晶衍射分析,对这些化合物进行了详细的结构表征。结果表明,这些化合物往往通过O-H…O,N-H…O及O-H…OC-M等氢键作用,形成一维或二维金属有机超分子结构。并且依赖于配体中羟甲基所处的不同位置,这些金属有机超分子表现出明显不同的结构特征。初步的催化活性测试表明,这些新化合物在苯乙炔三聚反应中具有明显的催化活性。
English
Syntheses and Catalytic Properties of Metal Carbonyl Derivatives with Hydroxymethyl Functionalized Pyrazoles
Abstract:
Reaction of tungsten or molybdenum carbonyl with 3(5)-hydroxymethyl-5(3)-methylpyrazole (L1), 4-hydroxymethylpyrazole (L2) and bis(3-hydroxymethyl-5-methylpyrazol-1-yl)methane (L3) yielded complexes LW(CO)5 (L=L1 or L2) and L3M(CO)4 (M=Mo or W), respectively. These complexes have been fully characterized by NMR, IR and X-ray crystal structural analyses, indicating that they form 1D or 2D organometallic supramolecular architectures through O-H…O, N-H…O and O-H…OC-M hydrogen bonding interactions, and these structures are significantly affected by the relative position of the hydroxymethyl group on the pyrazole ring. In addition, these complexes show moderate catalytic activity for the cyclotrimerization reaction of phenylacetylene.
-
Key words:
- N ligand
- / pyrazole
- / hydrogen bond
- / metal carbonyl complex
- / catalysis
-
0. Introduction
It is well-known that hydrogen bonds play important roles in the self-assembly of metal complexes to form supramolecular architectures[1-2]. Organometallic building blocks can also aggregate into supramole-cular structures through hydrogen-bonding interac-tions[3-4]. Metal carbonyls as hydrogen bond acceptors in organometallic crystals engineering have been observed in several systems, in which these metal carbonyl derivatives show interesting one to three-dimensional supramolecular structures[5-8]. One the other hand, derivatives of pyrazoles have been used extensively in bioinorganic, coordination chemistry and organometallic fields because of their versatile coordination behavior towards main group and trans-ition metals[9-11]. Among pyrazole derivatives, hydroxy-methyl functionalized pyrazole is an excellent candidate for the construction of supramolecular architectures, since it not only has multiple coor-dination modes but also can form regular hydrogen bonding by functioning as both a hydrogen-bonding donor and acceptor[12-13]. The group 6 metal carbonyl complexes are of great interest to scientists since they are widely applied in electron beam induced deposition technique as well as employed as catalysts in various organic synthesis[14-15]. In this paper, we report the reaction of hydroxymethyl functionalized pyrazoles (L) with group 6 metal carbonyls, which yields a series of LW(CO)5 and LM(CO)4 (M=Mo or W) derivatives with organometallic supramolecular stru-ctures through O-H…O, N-H…O and O-H…OC-M hydrogen-bonding interactions, and the preliminary catalytic activity of these corresponding complexes in the cyclotrimerization reaction of phenylacetylene.
1. Experimental
Solvents were dried and freshly distilled prior to use according to standard procedures. All reactions were carried out under an atmosphere of argon. NMR spectra were recorded on a Bruker 400 spectrometer using DMSO-d6 as solvent, and the chemical shifts were reported with respect to the reference (internal SiMe4 for 1H and 13C NMR spectra). IR spectra were recorded as KBr pellets on a Tensor 27 spectrometer. Elemental analyses were carried out on an Elementar Vario EL analyzer. Bis(3-hydroxymethyl-5-methylpy-razol-1-yl)methane was prepared by the published method[16]. All the other chemicals were analytical reagents and used as received.
1.1 Syntheses of 1 and 2
3(5)-Hydroxymethyl-5(3)-methylpyrazole (0.112 g, 1 mmol) was added to a solution of W(CO)5THF in THF, prepared in situ by the irradiation of a solution of W(CO)6 (0.359 g, 1 mmol) in THF (60 mL) for 8 h. The mixture was stirred and heated at reflux for 4 h. After the reaction was completed, the solvent was removed under a reduced pressure, and the residue was isolated by column chromatography on silica using ethyl acetate/hexane (1: 2, V/V) as the eluent to give 1 and 2 as yellow solids.
Complex 1: Yield: 12%. 1H NMR: δ 2.28 (s, 3H, CH3), 4.46 (d, J=5.6 Hz, 2H, CH2), 5.43 (t, J=5.6 Hz, 1H, OH), 6.20 (s, 1H, H4 of pyrazole), 13.28 (s, 1H, NH). 13C NMR: δ 16.4 (CH3), 54.5 (CH2), 105.6 (C4 of pyrazole), 148.3, 153.3 (C3 and C5 of pyrazole), 198.2 (4 CO), 202.5 (CO). IR(cm-1): ν(OH) 3 208; ν(NH) 3 150; ν(CO) 2 073, 1 918 (br), 1 879. Anal. Calcd. for C10H8N2O6W(%): C 27.55, H 1.85, N 6.42; Found(%): C 27.69, H 1.78, N 6.65.
Complex 2: Yield: 33%. 1H NMR: δ 2.29 (s, 3H, CH3), 4.47 (s, 2H, CH2), 5.45 (s, 1H, OH), 6.21 (s, 1H, H4 of pyrazole), 13.29 (s, 1H, NH). 13C NMR: δ 15.9 (CH3), 54.0 (CH2), 105.1 (C4 of pyrazole), 147.8, 152.8 (C3 and C5 of pyrazole), 197.7 (4 CO), 202.1 (CO). IR(cm-1): ν(OH) 3 234; ν(NH) 3 162; ν(CO) 2 073, 1 984, 1 915, 1 847. Anal. Calcd. for C10H8N2O6W(%): C 27.55, H 1.85, N 6.42; Found(%): C 27.62, H 1.94, N 6.29.
1.2 Synthesis of 3
The solution of 4-hydroxymethylpyrazole (49 mg, 0.5 mmol) and W(CO)6 (180 mg, 0.5 mmol) in THF (30 mL) was irradiated with a 300 W high-pressure mercury lamp for 8 h at room temperature. After the reaction was completed, the solvent was removed under a reduced pressure, and the residue was isolated by column chromatography on silica using ethyl acetate/hexane (1: 1, V/V) as the eluent to give 3 as a yellow solid. Yield: 125 mg (60%). 1H NMR: δ 4.37 (d, J=5.3 Hz, 2H, CH2), 5.01 (t, J=5.3 Hz, 1H, OH), 7.82 (s, 1H) and 7.86 (s, 1H) (H3 and H5 of pyrazole), 13.64 (s, 1H, NH). The signals at 5.01 and 13.64 disappeared when D2O was added. 13C NMR: δ 54.0 (CH2), 124.5 (C4 of pyrazole), 131.1, 146.4 (C3 and C5 of pyrazole), 198.3 (4CO), 202.8 (CO). IR(cm-1): ν(OH) 3 182; ν(NH) 3 137; ν(CO) 2 074, 1 968(sh), 1 909, 1 859. Anal. Calcd. for C9H6N2O6W(%): C 25.62, H 1.43, N 6.64; Found(%): C 25.28, H 1.25, N 6.38.
1.3 Synthesis of 4
Complex 4 was similarly obtained using 3, 5-dimethyl-4-hydroxymethylpyrazole instead of 4-hydro-xymethylpyrazole as above-mentioned synthesis of 3. Yield: 57%. 1H NMR: δ 2.23 (s, 3H, CH3), 2.26 (s, 3H, CH3), 4.25 (s, 2H, CH2), 4.69 (s, br, 1H, OH), 12.93 (s, 1H, NH). 13C NMR: δ 8.9 (CH3), 14.0 (CH3), 52.3 (CH2), 117.8 (C4 of pyrazole), 141.2, 151.9 (C3 and C5 of pyrazole), 197.7 (4 CO), 202.1 (CO). IR(cm-1): ν(OH) 3 199; ν(NH) 3 159; ν(CO) 2 072, 1 969 (sh), 1 908, 1 888. Anal. Calcd. for C11H10N2O6W(%): C 29.36, H 2.24, N 6.22; Found(%): C 29.31, H 2.31, N 6.24.
1.4 Synthesis of 5
Bis(3-hydroxymethyl-5-methylpyrazol-1-yl)methane (0.118 g, 0.5 mmol) was added to a solution of Mo(CO)5THF in THF, prepared in situ by the irradiation of a solution of Mo(CO)6 (0.132 g, 0.5 mmol) in THF (60 mL) for 8 h. The mixture was stirred and heated at reflux for 4 h. After the reaction was completed, the solvent was removed under a reduced pressure, and the residue was purified by column chromatography on silica using acetone/hexane (2: 3, V/V) as the eluent to give 5 as a yellow solid. Yield: 0.13 g (57%). 1H NMR: δ 2.48 (s, 6H, CH3), 4.67 (d, J=5.2 Hz, 4H, CH2), 5.49 (t, J=5.2 Hz, 2H, OH), 6.17(s, br, 1H, CH2N), 6.30 (s, 2H, H4 of pyrazole), 6.51 (s, br, 1H, CH2N). 13C NMR: δ 10.8 (CH3), 57.1 (CH2), 58.3 (CH2), 105.5 (C4 of pyra-zole), 142.4, 157.9 (C3 and C5 of pyrazole), 220.1 (CO). IR(cm-1): ν(OH) 3 393; ν(CO) 2 024, 1 921, 1 843. Anal. Calcd. for C15H16MoN4O6(%): C 40.55, H 3.63, N 12.61; Found(%): C 40.18, H 3.42, N 12.87.
1.5 Synthesis of 6
Complex 6 was similarly obtained using W(CO)6 instead of Mo(CO)6 as above-mentioned synthesis of 5. Yield: 53%. 1H NMR: δ 2.50 (s, 6H, CH3), 4.63 (s, 2H, CH2), 4.72 (s, 2H, CH2), 5.56 (s, 2H, OH), 6.14 (d, J=13.1 Hz, 1H, CH2N), 6.34 (s, 2H, H4 of pyrazole), 6.55 (d, J=13.1 Hz, 1H, CH2N). 13C NMR: δ 11.4 (CH3), 58.4 (CH2), 59.6 (CH2), 106.1 (C4 of pyrazole), 143.0, 158.7 (C3 and C5 of pyrazole), 211.9 (CO). IR(cm-1): ν(OH) 3 393; ν(CO) 2 016, 1 908, 1 840. Anal. Calcd. for C15H16N4O6W(%): C 33.86, H 3.03, N 10.53; Found(%): C 33.92, H 3.25, N 10.37.
1.6 Catalyzed cyclotrimerization of phenylacetylene
Phenylacetylene (0.12 mL, 1 mmol) and complexes 1~6 (x%, molar ratio) were charged in the reaction tube with 5 mL of toluene. After the reaction mixture was stirred at reflux for 9 h, the volatile materials were removed under reduced pressure. The residuals were purified by column chromatography on silica using CH2Cl2/hexane (1: 10, V/V) as the eluent to give the products, which were analyzed by 1H NMR[17].
1.7 Crystal structure determination
Green-yellow crystals of 1~3 suitable for X-ray analysis were grown by slow diffusion of hexane into their CH2Cl2 solutions at -18 ℃. While crystals of 6 were obtained through slow diffusion of hexane into the acetone solution. All intensity data were collected on a Rigaku Saturn CCD detector using Mo Kα radia-tion (λ=0.071 073 nm) at -160 ℃. Semi-empirical absorption corrections were applied using the Crystal-clear program[18]. The O(6) atom in 3 was disordered over two sites, with the occupancy factor of 0.5. The complex 6 crystallized with one acetone and one water molecules in the asymmetric unit. The structures were solved by direct methods and difference Fourier map using SHELXS of the SHELXTL package and refined with SHELXL[19] by full-matrix least-squares on F2. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were added geometrically and refined with riding model position parameters. A summary of the fundamental crystal data for these complexes is listed in Table 1.
Table 1

 DownLoad:
CSV
DownLoad:
CSV
Complex 1 2 3 6·CH3COCH3·H2O Formula C10H8N2O6W C10H8N2O6W C9H6N2O6W C18H24N4O8W Formula weight 436.03 436.03 422.01 608.26 Crystal size/mm 0.20×0.18×0.12 0.20×0.18×0.12 0.20×0.18×0.12 0.20×0.18×0.12 Crystal system Monoclinic Monoclinic Monoclinic Triclinic Space group P21/n C2/c P21/n P1 a/nm 0.705 16(16) 1.676 1(5) 0.702 15(16) 1.019 9(2) b/nm 1.679 8(3) 0.900 9(2) 1.775 7(4) 1.051 6(2) c/nm 1.094 9(3) 1.881 2(5) 0.991 7(2) 1.066 6(3) α/(°) 88.411(15) β/(°) 96.347(5) 111.968(4) 91.905(6) 87.754(14) γ/(°) 78.571(15) V/nm3 1.288 9(5) 2.634 3(13) 1.235 8(5) 1.120 2(4) Z 4 8 4 2 Dc/(g·cm-3) 2.247 2.199 2.268 1.803 θ range/(°) 3.06~27.53 3.056~27.481 3.08~25.019 3.11~27.57 F(000) 816 1 632 784 596 μ/mm-1 8.986 8.793 9.368 5.206 Measured reflection 16 325 13 841 10 613 12 336 Unique reflection (Rint) 2 954 (0.031 5) 3 009 (0.025 6) 2 176 (0.035 5) 4 973 (0.023 4) Observed reflection with [I≥2σ(I)] 2 811 2 926 1 994 4 867 Parameter 178 181 182 292 GOF 1.088 1.471 1.216 1.065 Fesiduals R1, wR2 0.012 2, 0.030 1 0.013 4, 0.041 6 0.032 0, 0.073 4 0.013 4, 0.034 0 CCDC: 1835473, 1; 1835474, 2; 1835475, 3; 1835476, 6.
2. Results and discussion
2.1 Syntheses of complexes 1~6
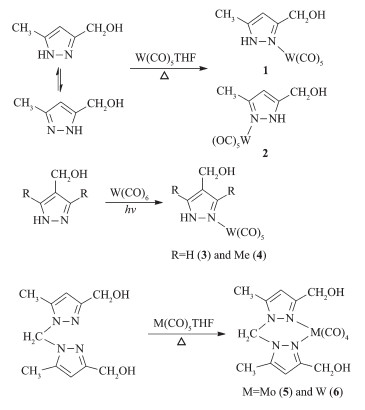
Reaction of 3(5)-hydroxymethyl-5(3)-methylpyra-zole with W(CO)5THF at refluxing THF or the direct photochemical reactions of 4-hydroxymethylpyrazoles with W(CO)6 at room temperature yielded complexes 1~4 (Scheme 1). Complexes 5 and 6 were also obtained through the similar reactions of bis(3-hydroxymethyl-5-methylpyrazol-1-yl)methane with M(CO)5THF (M=Mo or W) in moderate yields. Complexes 1~6 have been characterized by spectroscopic methods. Complexes 1~4 displayed similar IR spectra. These four complexes showed two characteristic absorption peaks for the O-H (3 199~3 234 cm-1) and N-H stretching bands (3 137 ~3 162 cm-1). A ν(C≡O) band at ca. 2 073 cm-1 corresponding to the A1eq mode for the pseudo C4v metal center in the M(CO)5 fragment[20] was observed in these four complexes, consistent with monodentate pyrazole complexes. The IR spectra of complexes 5 and 6 were different from those of 1~4. Four carbonyl absorption peaks in the range of 1 840~2 024 cm-1 were observed for 5 and 6, matching a typical cis-tetracarbonyl arrangement[21]. The NMR spectra of 1~6 also support the suggested structures. For example, the 13C NMR spectra of 1~4 showed two carbonyl carbon signals with ca. a 1: 4 intensity ratio, corres-ponding to a monosubstituted pentacarbonyl species. In addition, the protons of the methylene bridge of 5 and 6 displayed an AB system in their 1H NMR spectra, indicating that the inversion of the boat conformation of six-membered metallacycle (crystal structure of 6) was hindered possibly due to the repulsion among ligands.
Figure Scheme 1
 Figure Scheme 1. Syntheses of complexes 1~6
Figure Scheme 1. Syntheses of complexes 1~62.2 Crystal structures of complexes 1~3 and 6
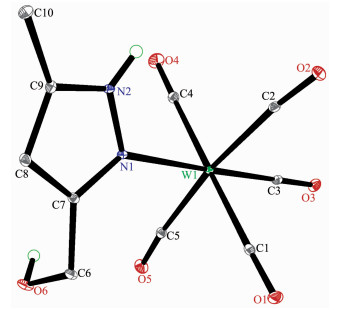
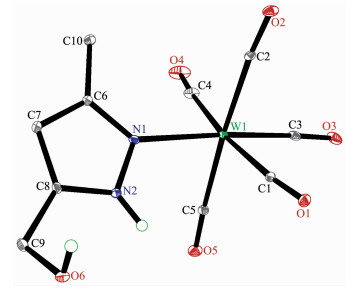
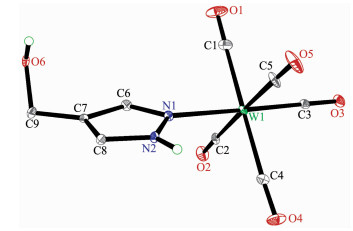
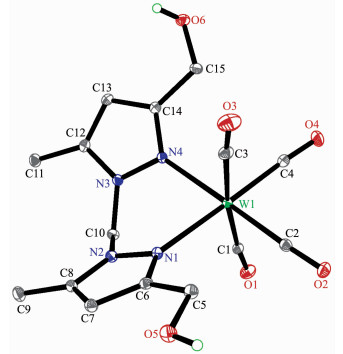
The structures of complexes 1~3 and 6 were further confirmed by X-ray crystallography, and are shown in Fig. 1~4, respectively. The selected bond distances and angles are listed in Table 2. Fig. 1~3 show that hydroxymethylpyrazoles coordinate to the metal center in a monodentate fashion in 1~3, causing them to possess a similar pentacarbonyl tungsten fragment, as shown by their NMR spectra. Complexes 1~3 also share some analogous structural parameters, such as similar W-N bond distance and N-W-C angle. The W-N distances in 1~3 (0.223 7~0.226 4 nm) are similar to those reported in other pentacarbonyl tungsten(0) complexes with azole ligands, such as 0.225 6(4) nm in CH2(3, 5-Me2Pz)(Bt-SnPh3)W(CO)5[22]. Fig. 4 shows that bis(3-hydroxymethyl-5-methylpyrazol-1-yl)methane acts as a chelating κ2-[N, N] bidentate ligand to the tungsten atom in 6, yielding a six-membered metallacycle with a boat conformation. The W-N bond distances (0.226 4(2) and 0.226 9(2) nm) are similar to those in 1~3, and comparable to those reported for other tetracarbonyl tungsten(0) derivatives with chelating bidentate pyrazolyl groups[23]. Additio-nally, two cis-carbonyls (C(2)O(2) and C(5)O(5) in 1, C(1)O(1) and C(4)O(4) in 2 and 3, as well as C(1)O(1) and C(3)O(3) in 6) are markedly distorted in these four complexes, as evidenced by the corresponding nonlinear C-W-C and W-C-O angles (Table 2), indi-cating the presence of steric repulsion between the ligand and these carbonyls.
Figure 1
 Figure 1. Molecular structure of 1 with 30% probability displacement ellipsoids
Figure 1. Molecular structure of 1 with 30% probability displacement ellipsoidsFigure 2
 Figure 2. Molecular structure of 2 with 30% probability displacement ellipsoids
Figure 2. Molecular structure of 2 with 30% probability displacement ellipsoidsFigure 3
 Figure 3. Molecular structure of 3 with 30% probability displacement ellipsoids
Figure 3. Molecular structure of 3 with 30% probability displacement ellipsoidsFigure 4
 Figure 4. Molecular structure of 6 with 30% probability displacement ellipsoids
Figure 4. Molecular structure of 6 with 30% probability displacement ellipsoidsTable 2
Table 2. Selected bond distances (nm) and angles (°) for complexes 1~3 and 6DownLoad:
CSV
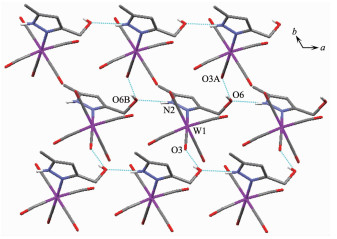
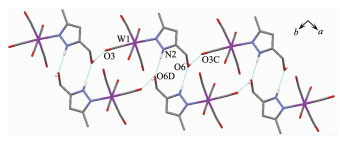
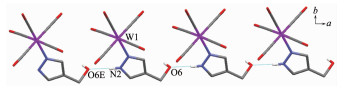
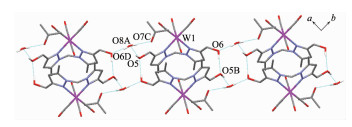
Complex 1 W(1)-N(1) 0.226 4(2) W(1)-C(1) 0.204 5(2) W(1)-C(3) 0.196 1(2) C(1)-O(1) 0.113 6(2) C(3)-O(3) 0.116 1(2) C(6)-O(6) 0.143 4(2) W(1)-C(2)-O(2) 173.0(2) W(1)-C(5)-O(5) 174.0(2) C(2)-W(1)-C(5) 172.0(1) Complex 2 W(1)-N(1) 0.225 3(2) W(1)-C(1) 0.2047(3) W(1)-C(3) 0.196 1(3) C(1)-O(1) 0.114 3(4) C(3)-O(3) 0.116 1(4) C(9)-O(6) 0.142 2(3) W(1)-C(1)-O(1) 173.7(2) W(1)-C(4)-O(4) 174.3(4) C(1)-W(1)-C(4) 172.3(1) Complex 3 W(1)-N(1) 0.223 7(6) W(1)-C(1) 0.202 8(8) W(1)-C(3) 0.198 0(7) C(1)-O(1) 0.113 0(10) C(3)-O(3) 0.115 9(9) C(9)-O(6) 0.147 1(15) W(1)-C(1)-O(1) 176.0(7) W(1)-C(4)-O(4) 175.6(7) C(1)-W(1)-C(4) 174.6(3) Complex 6 W(1)-N(1) 0.226 4(2) W(1)-N(4) 0.226 9(2) C(1)-O(1) 0.114 3(2) W(1)-C(1) 0.203 6(2) W(1)-C(4) 0.195 5(2) C(5)-O(5) 0.142 7(2) W(1)-C(1)-O(1) 174.1(2) W(1)-C(3)-O(3) 175.1(2) C(1)-W(1)-C(3) 172.95(7) N(1)-W(1)-N(4) 81.44(6) N(1)-W(1)-C(4) 177.74(6) N(2)-C(10)-N(3) 111.4(2) It is noteworthy that although complexes 1~3 have a similar molecular skeleton, they show significantly different supramolecular structures (Fig. 5~7) owing to the different substitutional position of hydroxymethyl on the pyrazolyl ring. For example, Fig. 5 shows that complex 1 aggregates into a 2D supramolecular network through O-H…O(carbonyl) and N-H…O(hydroxyl) hydrogen bonds, and Fig. 6 illustrates that complex 2 only extends into a 1D supramolecular double chain. Moreover, Fig. 7 shows that the metal carbonyls in 3 do not participate in the hydrogen bonding interac-tions, and this molecule is only linked into a one-dimensional chain via intermolecular N-H…O(hyd-roxyl) hydrogen bonds. In addition, Fig. 8 shows that complex 6 forms a 1D supramolecular double chain with macrocyclic units through crystallization water molecule, and also no hydrogen bond is observed between the metal carbonyls with hydroxyl protons.
Figure 5
 Figure 5. Two dimensional supramolecular structure of 1
Figure 5. Two dimensional supramolecular structure of 1Figure 6
 Figure 6. One dimensional supramolecular structure of 2
Figure 6. One dimensional supramolecular structure of 2Figure 7
 Figure 7. One dimensional supramolecular structure of 3
Figure 7. One dimensional supramolecular structure of 3Figure 8
 Figure 8. One dimensional supramolecular structure of 6·CH3COCH3·H2O
Figure 8. One dimensional supramolecular structure of 6·CH3COCH3·H2O2.3 Catalytic activity of complexes 1~6
The transition metal-catalyzed cyclotrimerization reaction of alkynes has been widely used to prepare various polysubstituted benzene derivatives in recent years[24]. Molybdenum carbonyl and its derivatives have exhibited efficient catalytic activity in the cyclotrimer-ization of alkynes[25-26], but it seems that phenylacety-lene tends to form polyphenylacetylene when tungsten carbonyl was used as the catalyst[27]. Herein, our preli-minary studies showed that all these molybdenum and tungsten carbonyl derivatives exhibited effective catalytic activity in the cyclotrimerization reaction of phenylacetylene (Table 3). Two isomers were obtained in moderate yields when 15%(n/n) of complex was used as the catalyst. 1, 3, 5-Trisubstituted benzene is the major product, similar to the result of the cyclotri-merization of phenylacetylene catalyzed by moly-bdenum carbonyl[26]. The ratio of isomers needs to be further improved in future work.
Table 3
Table 3. Catalytic activity for the cyclotrimerization of phenylacetyleneDownLoad:
CSV

Entry Cat. x/%(n/n) Yield (A+B)/%a Ratio of nA/nBb 1 1 15 63 1.4 2 2 15 66 1.4 3 3 5 43 1.5 4 3 15 63 1.5 5 3 20 62 1.5 6 4 15 56 1.5 7 5 15 53 1.5 8 6 15 55 1.5 a Isolated yield; b Determined by 1H NMR[17]. 3. Conclusions
In summary, a series of tungsten and molybde-num carbonyl derivatives with hydroxymethyl func-tionalized pyrazoles have been synthesized. These complexes show significantly different supramolecular structures owing to the different substitutional position of hydroxymethyl on the pyrazolyl ring. Preliminary catalytic studies prove that all these complexes exhibit moderate catalytic activity in the cyclotrimerization reaction of phenylacetylene.
-
-
[1]
Lewinski J, Zachara J, Justyniak I, et al. Coord. Chem. Rev., 2005, 249:1185-1199 doi: 10.1016/j.ccr.2004.11.013
-
[2]
Steiner T. Angew. Chem. Int. Ed., 2002, 41:48-76 doi: 10.1002/1521-3773(20020104)41:1<>1.0.CO;2-5
-
[3]
Mountford A J, Lancaster S J, Coles S J, et al. Organometallics, 2006, 25:3837-3847 doi: 10.1021/om060319d
-
[4]
Braga D, Grepioni F. Acc. Chem. Res., 2000, 33:601-608 doi: 10.1021/ar990143u
-
[5]
Xie Y F, Zhao D W, Tang L F. J. Organomet. Chem., 2016, 819:53-60 doi: 10.1016/j.jorganchem.2016.06.021
-
[6]
Braunstein P, Taquet J, Siri O, et al. Angew. Chem. Int. Ed., 2004, 43:5922-5925 doi: 10.1002/(ISSN)1521-3773
-
[7]
Camiolo S, Coles S J, Gale P A, et al. Chem. Commun., 2000:275-276
-
[8]
Yang P Y, Chang F C, Suen M C, et al. J. Organomet. Chem., 2000, 596:226-231 doi: 10.1016/S0022-328X(99)00546-X
-
[9]
Alkorta I, Claramunt R M, Díez-Barra E, et al. Coord. Chem. Rev., 2017, 339:153-182 doi: 10.1016/j.ccr.2017.03.011
-
[10]
Pettinari C, Tbcarub A, Galli S. Coord. Chem. Rev., 2016, 307:1-31 doi: 10.1016/j.ccr.2015.08.005
-
[11]
Otero A, Fernández-Baeza J, Lara-Sánchez A, et al. Coord. Chem. Rev., 2013, 257:1806-1868 doi: 10.1016/j.ccr.2013.01.027
-
[12]
Stumpf S, Wagner M, Bolte M. Acta Crystallogr. Sect. E:Struct. Rep. Online, 2004, E60:o1754-o1755
-
[13]
Yan G B. Acta Crystallogr. Sect. E:Struct. Rep. Online, 2007, E63:o2889
-
[14]
Kaushik M, Singh A, Kumar M. Eur. J. Chem., 2012, 3:367-394 doi: 10.5155/eurjchem.3.3.367-394.604
-
[15]
Ryan D E, Cardin D J, Hartl F. Coord. Chem. Rev., 2017, 335:103-149 doi: 10.1016/j.ccr.2016.12.018
-
[16]
Harit T, Bellaouchi R, Mokhtari C, et al. Tetrahedron, 2017, 73:5138-5143 doi: 10.1016/j.tet.2017.07.006
-
[17]
Breschi C, Piparo L, Pertici P, et al. J. Organomet. Chem., 2000, 607:57-63 doi: 10.1016/S0022-328X(00)00211-4
-
[18]
CrystalStructure 3. 7. 0, Crystalclear 1. 36, Crystal Structure Analysis Package, Rigaku and Rigaku/MSC, TX, 2000.
-
[19]
Sheldrick G M. Acta Crystallogr. Sect. A:Found. Crystallogr., 2008, A64:112-114
-
[20]
Kraihanzel C S, Cotton F A. Inorg. Chem., 1963, 2:533-540 doi: 10.1021/ic50007a028
-
[21]
Orgel L E. Inorg. Chem., 1962, 1:25-29 doi: 10.1021/ic50001a007
-
[22]
Yang H, Li S, Tang L F. Transition Met. Chem., 2016, 41:655-661 doi: 10.1007/s11243-016-0065-0
-
[23]
丁可, 孙遵明, 李厚谦, 等.无机化学学报, 2015, 31:345-352 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150219&journal_id=wjhxxbcnDING Ke, SUN Zun-Ming, LI Hou-Qian, et al. Chinese J. Inorg. Chem., 2015, 31:345-352 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150219&journal_id=wjhxxbcn
-
[24]
Saito S, Yamamoto Y. Chem. Rev., 2000, 100:2901-2915 doi: 10.1021/cr990281x
-
[25]
Ardizzoia G A, Brenna S, Lamonica L, et al. J. Organomet. Chem., 2002, 649:173-180 doi: 10.1016/S0022-328X(02)01114-2
-
[26]
刘宇宙, 周立山, 席婵娟.化学学报, 2006, 64:266-268 doi: 10.3321/j.issn:0567-7351.2006.03.015LIU Yu-Zhou, ZHOU Li-Shan, XI Chan-Juan. Acta Chim. Sinica, 2006, 64:266-268 doi: 10.3321/j.issn:0567-7351.2006.03.015
-
[27]
光善仪, 徐洪耀, 张胜义, 等.精细化工, 2000, 17:467-469 doi: 10.3321/j.issn:1003-5214.2000.08.011GUANG Shan-Yi, XU Hong-Yao, ZHANG Sheng-Yi, et al. Fine Chemicals, 2000, 17:467-469 doi: 10.3321/j.issn:1003-5214.2000.08.011
-
[1]
-
Figure 5 Two dimensional supramolecular structure of 1
Hydrogen bond distances: O(6)…O(3A) 0.290 6(2) nm, N(2)…O(6B) 0.279 8(2) nm; Symmetry codes: A: x+1/2, -y+1/2, z+1/2; B: x-1, y, z
Figure 6 One dimensional supramolecular structure of 2
Hydrogen bond distances: O(6)…O(3C) 0.293 5(3) nm, N(2)…O(6D) 0.279 8(2) nm; Symmetry codes: C: x+1/2, y-1/2, z; D:-x+2, -y+2, -z+1
Figure 7 One dimensional supramolecular structure of 3
Hydrogen bond distance: N(2)…O(6E) 0.270 6(2) nm; Symmetry codes: E: x-1, y, z
Figure 8 One dimensional supramolecular structure of 6·CH3COCH3·H2O
Hydrogen bond distances: O(5)…O(8A) 0.268 8(2) nm, O(6)… O(5B) 0.268 2(2) nm, O(8A)…O(7C) 0.275 4(2) nm, O(8A)… O(6D) 0.272 7(2) nm; Symmetry codes: A:-x, -y+2, -z+1; B: -x+1, -y+2, -z; C: -x, -y+1, -z+1; D: -x+1, -y+1, -z+1
Table 1. Crystallographic data and refinement parameters for complexes 1~3 and 6
Complex 1 2 3 6·CH3COCH3·H2O Formula C10H8N2O6W C10H8N2O6W C9H6N2O6W C18H24N4O8W Formula weight 436.03 436.03 422.01 608.26 Crystal size/mm 0.20×0.18×0.12 0.20×0.18×0.12 0.20×0.18×0.12 0.20×0.18×0.12 Crystal system Monoclinic Monoclinic Monoclinic Triclinic Space group P21/n C2/c P21/n P1 a/nm 0.705 16(16) 1.676 1(5) 0.702 15(16) 1.019 9(2) b/nm 1.679 8(3) 0.900 9(2) 1.775 7(4) 1.051 6(2) c/nm 1.094 9(3) 1.881 2(5) 0.991 7(2) 1.066 6(3) α/(°) 88.411(15) β/(°) 96.347(5) 111.968(4) 91.905(6) 87.754(14) γ/(°) 78.571(15) V/nm3 1.288 9(5) 2.634 3(13) 1.235 8(5) 1.120 2(4) Z 4 8 4 2 Dc/(g·cm-3) 2.247 2.199 2.268 1.803 θ range/(°) 3.06~27.53 3.056~27.481 3.08~25.019 3.11~27.57 F(000) 816 1 632 784 596 μ/mm-1 8.986 8.793 9.368 5.206 Measured reflection 16 325 13 841 10 613 12 336 Unique reflection (Rint) 2 954 (0.031 5) 3 009 (0.025 6) 2 176 (0.035 5) 4 973 (0.023 4) Observed reflection with [I≥2σ(I)] 2 811 2 926 1 994 4 867 Parameter 178 181 182 292 GOF 1.088 1.471 1.216 1.065 Fesiduals R1, wR2 0.012 2, 0.030 1 0.013 4, 0.041 6 0.032 0, 0.073 4 0.013 4, 0.034 0  下载: 导出CSV
下载: 导出CSV
Table 2. Selected bond distances (nm) and angles (°) for complexes 1~3 and 6
Complex 1 W(1)-N(1) 0.226 4(2) W(1)-C(1) 0.204 5(2) W(1)-C(3) 0.196 1(2) C(1)-O(1) 0.113 6(2) C(3)-O(3) 0.116 1(2) C(6)-O(6) 0.143 4(2) W(1)-C(2)-O(2) 173.0(2) W(1)-C(5)-O(5) 174.0(2) C(2)-W(1)-C(5) 172.0(1) Complex 2 W(1)-N(1) 0.225 3(2) W(1)-C(1) 0.2047(3) W(1)-C(3) 0.196 1(3) C(1)-O(1) 0.114 3(4) C(3)-O(3) 0.116 1(4) C(9)-O(6) 0.142 2(3) W(1)-C(1)-O(1) 173.7(2) W(1)-C(4)-O(4) 174.3(4) C(1)-W(1)-C(4) 172.3(1) Complex 3 W(1)-N(1) 0.223 7(6) W(1)-C(1) 0.202 8(8) W(1)-C(3) 0.198 0(7) C(1)-O(1) 0.113 0(10) C(3)-O(3) 0.115 9(9) C(9)-O(6) 0.147 1(15) W(1)-C(1)-O(1) 176.0(7) W(1)-C(4)-O(4) 175.6(7) C(1)-W(1)-C(4) 174.6(3) Complex 6 W(1)-N(1) 0.226 4(2) W(1)-N(4) 0.226 9(2) C(1)-O(1) 0.114 3(2) W(1)-C(1) 0.203 6(2) W(1)-C(4) 0.195 5(2) C(5)-O(5) 0.142 7(2) W(1)-C(1)-O(1) 174.1(2) W(1)-C(3)-O(3) 175.1(2) C(1)-W(1)-C(3) 172.95(7) N(1)-W(1)-N(4) 81.44(6) N(1)-W(1)-C(4) 177.74(6) N(2)-C(10)-N(3) 111.4(2)
下载: 导出CSV
-










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 3
- 文章访问数: 965
- HTML全文浏览量: 92
