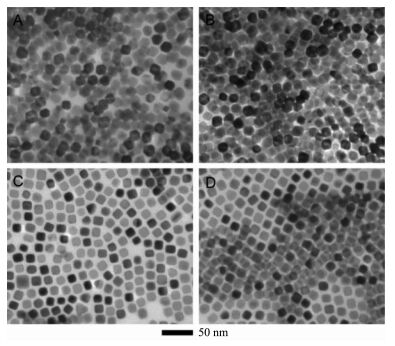
 图1
(A) Cu35Ni65正八面体, (B) Cu67Ni33正八面体, (C) Cu33Ni67纳米立方体和(D) Cu66Ni34纳米立方体的透射电镜图[24]
Figure1.
TEM images of (A) Cu35Ni65 octahedra, (B) Cu67Ni33 octahedra, (C) Cu33Ni67 nanocubes and (D) Cu66Ni34 nanocubes[24]
图1
(A) Cu35Ni65正八面体, (B) Cu67Ni33正八面体, (C) Cu33Ni67纳米立方体和(D) Cu66Ni34纳米立方体的透射电镜图[24]
Figure1.
TEM images of (A) Cu35Ni65 octahedra, (B) Cu67Ni33 octahedra, (C) Cu33Ni67 nanocubes and (D) Cu66Ni34 nanocubes[24]
引用本文:
徐兴良, 李莉萍, 张丹, 王彦, 李广社. 金属纳米复合催化剂合成与应用的最新进展[J]. 无机化学学报,
2017, 33(11): 1970-1990.
doi:
10.11862/CJIC.2017.257

Citation: XU Xing-Liang, LI Li-Ping, ZHANG Dan, WANG Yan, LI Guang-She. Recent Development on Synthesis and Application of Metal Nanocomposite Catalyst[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(11): 1970-1990. doi: 10.11862/CJIC.2017.257
Citation: XU Xing-Liang, LI Li-Ping, ZHANG Dan, WANG Yan, LI Guang-She. Recent Development on Synthesis and Application of Metal Nanocomposite Catalyst[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(11): 1970-1990. doi: 10.11862/CJIC.2017.257
金属纳米复合催化剂合成与应用的最新进展
摘要:
随着纳米技术的发展,金属纳米复合材料由于其特殊的物理化学性质和潜在的催化应用受到越来越多的关注。本文系统地介绍了金属纳米复合催化剂最新的研究进展。首先,我们介绍了一些金属纳米复合催化剂的合成方法。其次,为了更好的优化催化剂的结构与催化活性的关系,并深入理解催化反应机理,我们讨论了金属纳米复合催化剂一些重要因素(包括粒子粒径、形貌、组成、载体或配体)对催化活性和选择性的影响。最后,我们进一步介绍了金属纳米复合催化剂在还原不饱和化学键中的应用(N=O,N≡C和C=O),同时还对金属纳米复合催化剂的发展方向进行了展望。
English
Recent Development on Synthesis and Application of Metal Nanocomposite Catalyst
Abstract:
With the development of nanotechnology, metal nanocomposite materials have received extensive attention due to their unique physiochemical properties and potential catalytic applications. In this article, the latest development of metal nanocomposite catalysts is systematically introduced. First, we present some important synthetic approaches to the metal nanocomposite catalyst. Secondly, in order to better optimize the structure-catalytic activity correlations and understand the catalytic mechanisms, we discuss the influence of some critical factors (e.g. particle size, shape, composition and support or ligand) of metal nanocomposite catalyst on catalytic activity and selectivity. Finally, we pay special attention to metal nanocomposite catalyst for the reduction of unsaturated bonds (N=O, N≡C and C=O), and the research trends of metal nanocomposite catalyst are prospected.
-
0 引言
催化是当今化学品、燃料、材料、医药和食品生产及环境净化的支柱技术,是现代化学工业的基石[1-2]。催化科学的重要目的就是在分子层面理解催化反应和设计具有特定反应活性和选择性的催化剂。随着纳米科学和纳米技术的发展,纳米材料具有较小的粒径和更多的表面活性位点,能够更好的与反应底物接触,展现出优异的催化性能。其中,金属纳米粒子作为一个最重要的无机材料,相比其体相材料,显示出优异的电、磁、光等物理特性。金属纳米粒子受到了各个研究领域的广泛关注,特别是在各种催化反应中[3]。
在催化反应中,单一的金属纳米粒子作为重要的催化活性中心,由于具有较高的表面能,在制备过程和催化反应中可能发生团聚和失活,影响催化活性和选择性,降低使用寿命[4-5]。为了提高金属纳米粒子的催化性能,越来越多的研究者致力于在金属纳米催化剂中引入其他组分制备合成金属纳米复合催化剂[6-7]。一方面,通过其他组分的引入,由于存在电子效应,锚定效应等有助于提高金属纳米粒子的稳定性和分散性[8-9]。另一方面,金属纳米复合催化剂中各组分之间(如:金属与载体等)存在着较强相互作用,可以明显提高催化反应活性和选择性[10-11]。另外,通过构筑特殊结构的金属纳米复合催化剂可以提高金属纳米粒子的稳定性。比如核壳结构可以有助于金属纳米粒子的分散和阻止其团聚[12]。然而,合成高活性,高稳定性的金属纳米复合材料仍然面临着巨大的挑战。
近年来,人们更多的关注于精确可控合成金属纳米复合催化剂[2, 13]。纳米材料新的合成方法和技术的发展,可以更好的指导我们合成具有特定结构和形貌的金属纳米复合催化剂,推动纳米材料和纳米科学的进步。另外,由于金属纳米复合催化剂的催化性能往往受到粒径大小,形貌,结构和组成等各种因素的影响,因此系统的研究催化剂中的各种因素与催化性能之间的关系,可以为更好的理解和优化催化剂的结构与催化活性的关系以及研究催化机理提供实验和理论支持。基于以上讨论,本文首先系统介绍并评述近几年来金属纳米复合材料催化剂合成和制备的最新进展;然后详尽介绍金属纳米复合催化剂中尺寸,形貌,以及各组分之间的相互作用等对催化性能的影响;最后,介绍了金属纳米复合材料在还原不饱和化学键(还原N=O,N≡C和C=O)中的应用,并对其进行系统总结于分析。同时,本文还根据金属纳米复合催化剂的研究现状提出该领域面临的问题和发展趋势。
1 金属纳米复合材料的合成
纳米材料的合成与制备是纳米科学和纳米技术发展的基础。为了获得特定结构和性能的金属纳米复合材料,很多因素需要被考虑,比如每个组分的晶体结构,表面能,电子效应,配体封装等[2, 14]。近几年,金属纳米复合材料的合成取得了很大的进展。许多课题组和研究者通过各种方法合成了具有特定结构和形貌的金属纳米复合材料,并发展一些新的方法和技术。
1.1 化学还原法
化学还原法是合成金属纳米催化剂最常见方法。金属盐溶解于水或有机溶剂中,在表面活性剂和还原剂的作用下,经过一定反应条件可得到金属纳米颗粒。其中硼氢化钠作为一种强还原剂能够迅速还原金属离子特别是贵金属,使其快速成核形成金属纳米粒子[15-16]。Gu等[17]以PVP为保护剂,硼氢化钠为还原剂,获得了胶体金纳米粒子高度分散在氧化锰纳米棒上,金纳米粒子的平均粒径为1.9 nm。然而,强还原剂的使用会导致反应速率过快,金属纳米粒子形貌难以控制,且容易团聚。为了更好的控制反应速率,一些温和的还原剂,比如乙二醇[18],柠檬酸/钠[19],抗坏血酸[20],水合肼[21],甲酸[22]等被应用于合成金属纳米复合材料,尤其可以促使形成核壳结构的金属纳米复合材料。Gupta等[23]以抗坏血酸为还原剂,以硝酸铁和氯金酸为原料,在室温条件下合成了Fe@Au纳米粒子。将Fe@Au纳米粒子负载在氧化石墨烯上,在芳香族硝基化合物的还原过程中展现出较高的催化活性。
在化学还原法中,还原剂的选择决定了金属纳米复合催化剂的形貌和性能。除了上述传统的还原剂,最近一些新型的还原剂也得到了广泛的研究。Wang等[24]利用富氢试剂吗啉硼烷作为还原剂合成了具有可控形貌和特定元素比例的铜镍合金纳米粒子。这主要归因于吗啉硼烷在分解过程中快速产氢可以使金属离子快速成核。由于吗啉硼烷分解过程中产生的化学物质(氢气、吗啉)起到了封端剂的作用,并选择性吸附在金属纳米粒子的(110)晶面上,使这些晶面趋于热力学稳定。将吗啉硼烷的用量由20 mg增加到40 mg,导致出现了更多的(110)晶面,进而使铜镍合金的形貌由正八面体转化为立方体(图 1)。另外,合金中铜镍的比可以通过初始投料比改变,并且投料比与最终比是一致的,也没有发生相分离。该研究工作对利用化学还原法设计合成组分和形貌可控的非贵金属纳米材料提供了一个新的方法。
图1
(A) Cu35Ni65正八面体, (B) Cu67Ni33正八面体, (C) Cu33Ni67纳米立方体和(D) Cu66Ni34纳米立方体的透射电镜图[24]
Figure1.
TEM images of (A) Cu35Ni65 octahedra, (B) Cu67Ni33 octahedra, (C) Cu33Ni67 nanocubes and (D) Cu66Ni34 nanocubes[24]
目前,在利用化学还原法合成金属纳米复合材料的过程中,绝大部分都是在合成过程中加入还原剂和表面活性剂,这种合成方法面临着还原剂和表面活性剂除去和分离的问题。为了解决使用外部还原剂的问题,我们课题组在水热条件下,利用化学反应中歧化反应合成一系列的Sn的金属间化合物(Sn-Cu,Sn-Ni,Sn-Co)[25]。以Sn-Cu为例,反应机理如图 2所示:(1)在加热之前,由于Sn2+在碱性溶液中是以SnO22-的形式存在,具有较高的还原性,Cu2+被还原为Cu2O;(2)在水热反应过程中,由于温度和压力的增加,SnO22-的还原能力增强,继续将Cu2O还原为金属Cu;(3) Cu2O/Cu的还原电势(-0.360 V)要高于[SnO2]2-/Sn的还原电势(-0.909 V),SnO22-发生歧化反应生成Sn单质和[SnO3]2-,其中Sn单质原位与铜单质结合形成Cu3Sn金属间化合物。在原位合成中引入无机自歧化反应将为还原反应和其他金属复合材料的合成提供一个全新的思路。
1.2 热分解法
通过高温热分解有机金属化合物被广泛应用于合成金属纳米粒子及其复合物[26-27]。该方法通常在惰性条件下进行。相比于其他方法,该方法可以很好的控制纳米粒子的粒径,改变各种组分的比例,能够得到在液相条件下无法合成的金属纳米复合材料。
在热分解过程中,油胺类化合物被广泛使用。一方面,油胺的NH2基团可以与金属离子形成微弱的结合力,加上长的烷链基团,能够很好的控制金属纳米粒子的大小,提高稳定性,还可以改变纳米粒子的表面结构[28-29]。另一方面,在高温条件下,油胺作为一种弱的还原剂,可以用于合成金属纳米粒子和金属氧化物。Yu等[30]通过油胺的作用,在300 ℃热分解乙酰丙酮钴和乙酰丙酮铂合成了CoPt纳米粒子。在制备过程中油胺起到了表面活性剂、溶剂和还原剂的作用。CoPt纳米粒子的粒径和形貌可以很好的被控制(图 3),并且Co和Pt的比例可以通过前驱体的加入量控制。该制备方法能够很好的控制纳米粒子的成核和生长过程,可以优化纳米粒子的结构和表面性能。Niu等[31]以乙酰丙酮镍,乙酰丙酮铂和六羰基钨(W(CO)6)为原料,溶解于油酸,油胺和二苄醚的混合溶液,然后注入聚四氟反应管中。在高温作用下,由于W(CO)6产生一氧化碳气体,可以确保液滴源源不断的形成。液体作为一个微小的“反应堆”应用于Pt-Ni八面体的成核与生长。Pt-Ni八面体的粒径可以通过改变油酸和油胺的组成调控,此外Pt-Ni八面体的中的Pt-Ni比例能够通过乙酰丙酮铁的添加量改变。
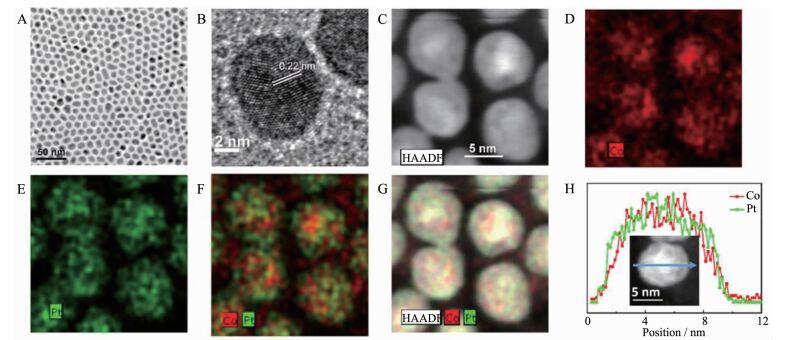 图3
(A) 9.5 nm Co47Pt53 NPs的透射电镜图; (B)显示在(A)中典型的纳米粒子的高分辨透射电镜图; (C) Co47Pt53 NPs的高角度环形暗场图; (D~F) Co(红色)/Pt(绿色)的元素分布; (G) Co47Pt53 NPs高角度环形暗场图和相应的元素分布; (H) Co47Pt53 NP的EDS图[30]
Figure3.
(A) TEM image of the 9.5 nm Co47Pt53 NPs; (B) HRTEM image of a representative NP shown in (A); (C) HAADF image of four representative Co47Pt53 NPs; (D~F) Elemental mappings of Co (red)/Pt (green) signals combined (F) and single element Co (red) (D) and Pt (green) (E); (G) HAADF image and the corresponding elemental map of four representative Co47Pt53 NPs; (H) EDS line scan crossing a Co47Pt53 NP; Inset shows the NP scanned[30]
图3
(A) 9.5 nm Co47Pt53 NPs的透射电镜图; (B)显示在(A)中典型的纳米粒子的高分辨透射电镜图; (C) Co47Pt53 NPs的高角度环形暗场图; (D~F) Co(红色)/Pt(绿色)的元素分布; (G) Co47Pt53 NPs高角度环形暗场图和相应的元素分布; (H) Co47Pt53 NP的EDS图[30]
Figure3.
(A) TEM image of the 9.5 nm Co47Pt53 NPs; (B) HRTEM image of a representative NP shown in (A); (C) HAADF image of four representative Co47Pt53 NPs; (D~F) Elemental mappings of Co (red)/Pt (green) signals combined (F) and single element Co (red) (D) and Pt (green) (E); (G) HAADF image and the corresponding elemental map of four representative Co47Pt53 NPs; (H) EDS line scan crossing a Co47Pt53 NP; Inset shows the NP scanned[30]
另外,热分解方法也应用于其他贵金属纳米复合材料和非贵金属复合材料的合成。Lü等[26]以乙酰丙酮镍和氯金酸为原料,在油胺和少量油酸的作用下成功合成了粒径和组分可控的单分散的NiAu合金纳米粒子。该方法不仅局限于NiAu合金纳米粒子的合成,也可以扩展到FeAu和CuAu合金纳米粒子。在热分解过程中,通过改变金属前驱体和形貌控制剂,可以合成出不同结构的金属纳米复合材料。Zeng等[32]成功的合成了Cu-ZnO核壳纳米粒子(图 4),具体的制备方法如下:一定量的乙酰丙酮铜、三辛基氧化膦和1,2-十二烷二醇溶于油胺和二苄醚的混合溶液中,70 ℃加热10 min,再升温到205 ℃保持15 min,然后再加入乙酰丙酮锌、油胺以及二苄醚的混合溶液,最后265 ℃保持30 min。样品冷却至室温,离心洗涤。作者通过改变铜前驱体的种类以及不同的形貌控制剂,成功合成了不同形貌的Cu-ZnO纳米复合材料,比如纳米多枝形,纳米棱锥,核壳纳米线。该合成策略为合成不同形貌的金属纳米复合材料提供一个新的方法。
1.3 微乳法
微乳法通常是指2种互不相溶的溶剂在表面活性剂的作用下形成微乳液,金属离子在微乳液中经过成核、生长、热处理后得纳米粒子[33-34]。微乳液法是制备单分散纳米粒子的重要手段,近年来得到了很大的发展和完善。与传统的制备方法相比,微乳法展现了良好的粒子单分散和界面性。Roberto等[35]在水/油微乳液体系中(3%水/16.5%聚氧乙烯月桂醚(BRIJ30)/80.5%正庚烷),用硼氢化钠还原氯铂酸钾得到Pt纳米粒子。为了获得了最多的立方体结构,提高其电化学性能。作者研究了在微乳液中加入不同量的盐酸对立方体的Pt纳米粒子表面结构产生的影响。如图 5所示,盐酸的加入量是影响Pt纳米粒子的形貌和表面结构的关键因素。当盐酸的量为25%时,获得了最多的立方体结构,其电化学性能是不加盐酸的6倍。该课题组也研究了H2SO4的加入量对Pt纳米粒子的形貌和表面结构的影响[36]。另外,改变水油相各组分的比例也会影响催化剂的性能。李峰等[37]采用十六烷基三甲基溴化铵(CTAB)/正丁醇/环己烷/H2PtCl6溶液的微乳液体系制备了非晶态Pt-B/Al2O3催化剂。考察了微乳液体系中CTAB/正丁醇/环己烷质量比、微乳液体系中水相含量对催化剂加氢性能的影响。
为了获得具有特殊结构的金属纳米复合催化剂,Mitsudome等[38]将微乳法与氧化还原-共沉淀法结合成功制备了Au@CeO2核壳结构的纳米粒子。具体方法如下:将氯金酸和硝酸铈的溶液与聚氧代乙烯(5)壬基苯基醚和环己烷混合,命名为A溶液;同时氢氧化钠溶液与聚氧代乙烯(5)壬基苯基醚和环己烷混合,命名为B溶液。将A和B两个溶液混合,并在室温下搅拌30 mim,乙醇加入到上述混合溶液。溶液离心获得的沉淀物洗涤干燥焙烧得到Au@CeO2。如图 6a所示Au@CeO2的直径为20 nm,在图 6b中可以看出,高电子密度的金核直径为8.6 nm,CeO2壳为2 nm。由于核壳结构的存在,最大化地暴露了金纳米粒子核与CeO2壳界面之间的活性位点,使其在半氢化各种炔烃中显示出优异的选择性。
1.4 化学气相沉积
化学气相沉积是利用气态或蒸汽态的物质在气相或者气固界面上反应生成固态沉积物的技术。该方法主要不仅应用薄膜的生长,而且在纳米材料的合成中也得到了广泛的应用[39-40]。通过对气相沉积过程中各种参数进行调控(如:基底、前驱体、生长温度、生长时间、压力等)可以成功制备出高质量金属纳米复合材料。宋春蕊等[41]通过化学气相沉积法,以乙酰丙酮钴为原料,在600~800 ℃温度下制备碳包覆钴的核/壳结构Co/C纳米颗粒。Co/C纳米颗粒中Co的尺寸在10~60 nm之间,C壳层的厚度在10~20 nm之间。该课题组采用化学气相沉积法还合成了Cu/C,Ni/C和Fe/C的核/壳结构纳米颗粒,并系统的研究了其形成机理[42-44]。在最近几年化学气相沉积也有了很多改进,比如激光诱导化学气相沉积,等离子体增强化学气相沉积等。另外原子沉积也是化学气相沉积法一种改进方法,原子沉积可以获得均一的高度分散的金属纳米粒子并能够很好的沉积到多孔基质上[45-46]。这种方法对于金属纳米粒子,特别是负载型催化剂在催化反应的应用是十分重要的[46]。Jiang等[47]利用原子沉积的方法,实现了镍纳米粒子高度分散到多孔的二氧化硅上(Ni/SiO2)。其中镍的粒径小于3 nm,并且Ni/SiO2在芳香族硝基化合物的还原中表现出较高的反应活性和选择性。
1.5 其他方法
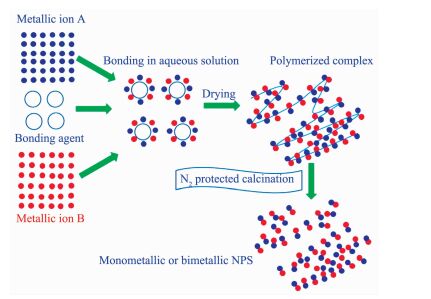
在传统的金属纳米粒子材料的合成过程中,往往会使用一些价格昂贵的还原剂和有毒的有机试剂。因此,发展绿色环保的合成方法显得尤为重要。Li等[48]以来源丰富的生物质衍生物(如可溶性淀粉、蔗糖等)为粘合剂,可以与金属离子形成网状结构的高分子金属配合物(图 7)。该网状的配合物在氮气气氛下热分解制备出Cu-Pt、Ni-Pt、Ni-Pd、Pt、Pd、Ag、Cu、Ni等金属纳米粒子。通过各种检测手段,作者进一步解释了金属纳米粒子的形成机理。金属纳米粒子的制备分为2个阶段。第一个阶段,高分子金属配合物在147~277 ℃之间在氮气氛围下热分解产生H2、CH4、H2O、NH3和CO2等气体;第二个阶段,在327~427 ℃之间,C-O官能团消除,配合物继续分解。在H2和CH4还原气氛下,晶核形成和不断生长,最后形成金属纳米粒子。该方法是以生物质衍生物为粘合剂和碳源,通过高温热分解还原金属离子合成金属纳米粒子及其合金,进而扩展了金属纳米复合材料合成的方法,并且符合绿色可持续发展的理念。
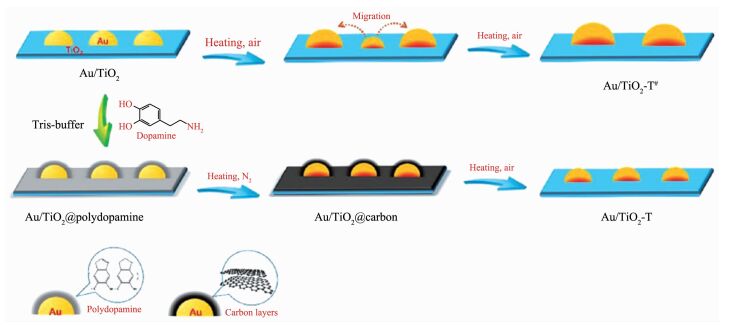
众所周知,金属纳米粒子具有较低的塔曼温度和高表面能,导致其热稳定性较低,在催化反应中容易发生团聚和失活的现象[49]。一直以来提高金属纳米粒子的稳定性是金属纳米复合材料合成方法发展的趋势之一。很多课题组和研究工作者为此做了大量的工作。Zhan等[50]创造性的提出了一种在高温氧化过程中稳定金属纳米粒子的合成策略(图 8),具体方法是:以Au/TiO2为例,第一步,在Au/TiO2制备过程中引入多巴胺,由于多巴胺具有优异的黏附性,使得多巴胺在Au纳米粒子和TiO2载体表面形成有机层;第二步,在氮气氛围下焙烧将有机层转变成碳层,在碳化过程中由于碳层的存在可以有效的隔离金纳米粒子,进而阻止其被烧结,并且粒径可以保持不变。其中更重要的是,Au纳米粒子和TiO2载体之间的相互作用显著增加;最后,在空气中进行高温焙烧,碳层消失从而暴露出金属活性中心。由于在氮气焙烧过程中Au纳米粒子和TiO2载体之间的相互作用得到加强,因此在空气高温焙烧过程中,Au纳米粒子发生烧结的可能性大大降低。该方法条件对其他贵金属(Pd和Pt)也有很好的适用性,具有一定的普适性。
其他的制备金属纳米复合材料的方法还有很多,比如沉淀法、水热合成法、溶胶凝胶法等。限于篇幅这里不再一一介绍。
2 金属纳米复合催化剂结构与催化性能的关系
随着纳米材料合成方法的不断发展和创新,金属纳米复合催化剂的结构可以很好的控制,能够帮助我们更加准确的研究催化剂中各个因素与催化性能之间的关系。很多研究结果表明金属纳米复合催化剂高的催化活性和选择性不仅仅与比表面积和表面活性位点有关,还受到尺寸,形貌,以及各种组分的影响[3]。在金属纳米复合材料中,金属纳米粒子是主要的纳米分散相,可以通过调节粒径和形貌来影响催化性能。另外,金属纳米复合材料的一个独特的性质是能够将各种组分的功能集成到一个单一的单元中,金属纳米复合材料可以展现不同于金属纳米粒子和简单物理混合的特殊性质,克服单一组分在催化反应中的问题和不足。因此研究催化剂中各种因素与催化活性之间的关系,对于发展高效的纳米催化剂和更好的理解催化机理尤为重要。
2.1 尺寸因素
在各种因素中,首先提到是纳米催化剂的粒径大小。纳米催化材料的电子结构受到其粒径大小的影响,使得纳米材料与常规材料在化学和物理性能上有着显著的不同[51]。例如体相金是化学惰性的,而金纳米粒子却能够显示出优异的催化性能[52]。随着纳米材料粒径的减小,表面原子数迅速增加。由于表面原子周围缺少相邻的原子,有许多悬空键,具有不饱和性,易与其他原子相结合而稳定下来,故表现出很高的化学活性[53]。随着粒径的减小,纳米材料的比表面积、表面能及表面结合能都迅速增大,有利于降低催化反应中的能垒[54]。研究纳米催化剂的粒径与其催化性能之间的关系一直以来是催化剂设计的重要内容。
近年来,很多工作都致力于研究催化剂的尺寸与其催化性能之间的关系。喹啉选择性加氢反应是一类重要的工业反应[55],贵金属纳米粒子在该反应中往往表现出较低的催化活性,使得该反应通常在比较苛刻的条件才能进行。为了降低反应条件,Gao等[56]合成了超小的、尺寸能够精确调控的铂纳米粒子(~1 nm)。在喹啉选择性加氢反应中,粒径与催化性能呈“火山型”构型(如图 9c),即,1.2 nm的铂纳米粒子较于其它的尺寸(0.7,2.4,5.3 nm)具有最佳的催化活性,并且能够使该反应在常温常压下反应。主要原因是当粒径大于1.2 nm,铂纳米粒子与反应物的相互作用比较弱;当小于1.2 nm,相互作用比较强。1.2 nm铂纳米粒子反应5次后仍然保持较高的活性和选择性(图 9d),并没有发生分解和团聚。铂纳米粒子的尺寸效应归因于纳米粒子的d带电子结构以及贵金属纳米粒子与反应物和产物之间的相互作用。该工作揭示了贵金属纳米催化剂尺寸效应对有机加氢反应的影响,并对尺寸效应的作用机理提出了新的解释,另一方面也为高活性和高选择性的金属纳米催化剂的设计提供了新的策略。Li等[57]也系统讨论了钯纳米粒子的尺寸对甲酸分解的影响。作者合成5种粒径不同的钯纳米粒子负载在碳上,从2.1到4.5 nm。实验结果显示了随着粒径的减小,甲酸的分解能力显著增强(图 10)。其中粒径为2.1 nm的钯纳米颗粒复合催化剂的TOF是4.5 nm的3.6倍。小尺寸的钯纳米粒子高的催化活性源于较高的分散度和高比例的带正电性的钯物种。通过库仑效应,带正电性的钯物种与带负电性的甲酸盐物种的结合能力增强,从而促进反应的进行。
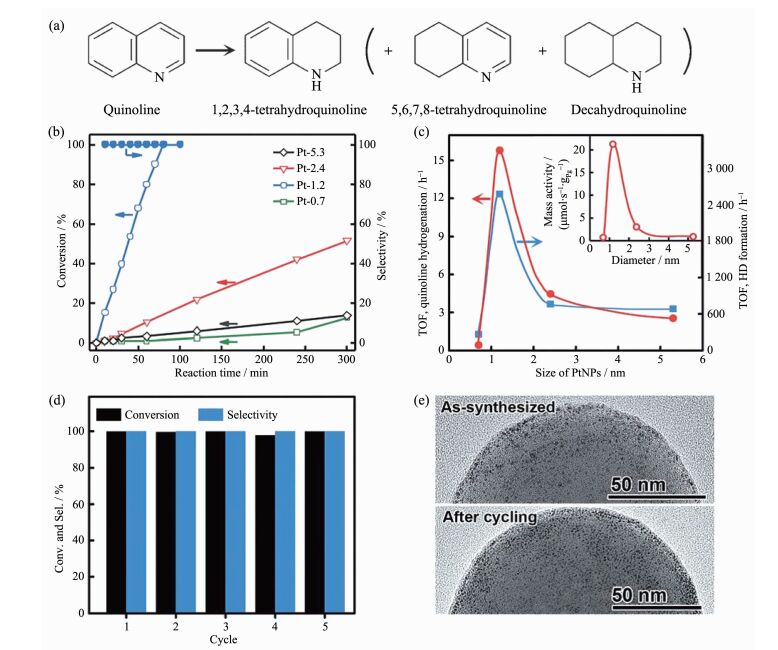 图9
(a) 喹啉的加氢产物; (b)用不同粒径Pt作为催化剂, 喹啉的转化率和1, 2, 3, 4-四氢喹啉选择性随时间变化的曲线; (c)TOF随粒径Pt变化的曲线(每个表面积的Pt); 活化H2和D2的TOF比较, (d) Pt-1.2反应5次的转化率和选择性的比较; (d)反应前后催化剂TEM电镜图[56]
Figure9.
(a) Possible products of the quinoline hydrogenation; (b) Plots of conversion of quinoline and selectivity toward 1, 2, 3, 4-tetrahydroquinoline against reaction time with Pt-x as the catalyst; (c) Plot of TOF (per surface Pt atom) against diameter of the PtNPs. The TOF for activation of H2 and D2 to form HD is listed for comparison. Inset: Size-dependent mass activity (per unit mass of Pt) of the PtNPs in hydrogenation of quinoline; (d) Change of conversion and selectivity in 5 cycles of the catalysis with Pt-1.2 as the catalyst; (e) TEM images of the catalyst before and after cycling[56]
图9
(a) 喹啉的加氢产物; (b)用不同粒径Pt作为催化剂, 喹啉的转化率和1, 2, 3, 4-四氢喹啉选择性随时间变化的曲线; (c)TOF随粒径Pt变化的曲线(每个表面积的Pt); 活化H2和D2的TOF比较, (d) Pt-1.2反应5次的转化率和选择性的比较; (d)反应前后催化剂TEM电镜图[56]
Figure9.
(a) Possible products of the quinoline hydrogenation; (b) Plots of conversion of quinoline and selectivity toward 1, 2, 3, 4-tetrahydroquinoline against reaction time with Pt-x as the catalyst; (c) Plot of TOF (per surface Pt atom) against diameter of the PtNPs. The TOF for activation of H2 and D2 to form HD is listed for comparison. Inset: Size-dependent mass activity (per unit mass of Pt) of the PtNPs in hydrogenation of quinoline; (d) Change of conversion and selectivity in 5 cycles of the catalysis with Pt-1.2 as the catalyst; (e) TEM images of the catalyst before and after cycling[56]
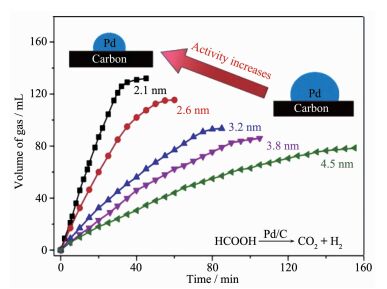 图10
不同粒径Pt随时间变化甲酸产生气体的量(CO2+H2)的变化趋势
Figure10.
Plots of the volume of the generated gas (CO2+H2) versus time with five different particle sizes
图10
不同粒径Pt随时间变化甲酸产生气体的量(CO2+H2)的变化趋势
Figure10.
Plots of the volume of the generated gas (CO2+H2) versus time with five different particle sizes
除了催化剂的粒径对化学反应有着重要的影响,Reske等[58]研究了铜纳米粒子的粒径与电化学还原CO2之间的关系。铜纳米粒子粒径的改变影响了电化学还原CO2的产物的分布。随着铜纳米粒子粒径的降低,特别是粒径小于5 nm时,对于H2和CO的选择性明显增强,碳氢化合物(甲烷和乙烯)的选择性降低。铜纳米粒子粒径的改变会影响铜物种表面配位数,从而影响铜物种对CO2电化学还原过程中中间物种的吸附和键合能力。当粒径小于2 nm,铜物种的表面配位数小于8。这些低配位数的活性位点加速了H2的析出,用于还原CO2到CO。小尺寸的铜纳米粒子不利于继续还原CO制备碳氢化合物。一种合理的解释是中间物种(CO和H)在小尺寸铜纳米粒子表面有着较强的结合能力,不利于中间物种继续结合和反应生成碳氢化合物。催化活性-选择性-尺寸的关系对金属纳米催化剂电化学还原CO2提供了新的解释。
2.2 形貌因素
由于金属纳米粒子不同的暴露面对于催化反应中活化能的降低存在着差异[59-60],因此金属纳米复合材料的形貌极大的影响其催化性能。另外金属上不同暴露面的活性位点可以优先或者选择性吸附/解离反应分子,包括基底、中间体和最终产物,最终影响其反应速率和反应途径[61]。
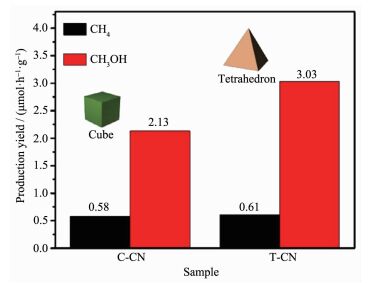
Bratlie等[62]研究了Pt的不同形貌对于苯加氢反应的影响。立方相和立方八面相的催化性能明显不同。立方相Pt可以催化还原苯到环己烷,立方八面体相可以得到环己烷和环己烯(图 11a)。这种选择性的差异是因为立方相Pt由(100)晶面组成,而立方八面体相由(100)和(111)晶面组成,其中(100)面有利于环己烷的生成,(111)面有利于环己烯的生成。Cao等[63]研究了g-C3N4表面上Pd助催化剂不同晶面对光还原CO2的影响。通过改变Pd的(111)和(100)晶面的数量,改变助催化剂的表面结构,进而影响载流子转移、CO2的吸附和CH3OH的脱附。从图 12中可以看出四面体相Pd暴露的(111)晶面对光催化还原CO2到CH3OH的催化活性明显优于立方相Pd暴露的(100)晶面。根据理论计算和实验结果,作者认为四面体相Pd暴露的(111)晶面更有利于CO2的吸附和CH3OH的脱附。这个工作可以更好的理解金属助催化剂表面结构的改变与光催化性能之间的关系。
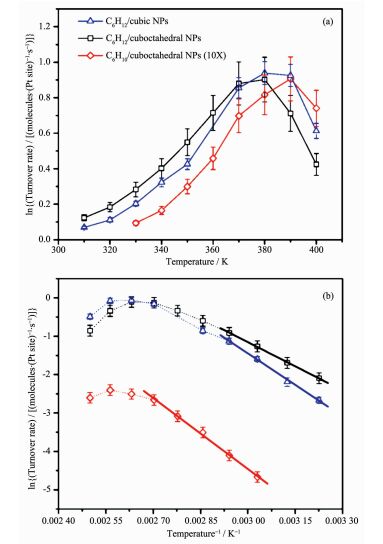 图11
(a) 在1.3×104 Pa C6H6, 1.3×105 Pa H2和8.5×105Pa Ar的条件下, 十四烷基三甲基溴化铵(TTAB)稳定的Pt立方相和Pt立方八面相对环己烷和环己烯的转换率; (b)相应的阿伦尼乌斯图[62]
Figure11.
(a) Turnover rates of cyclohexane (C6H12) and cyclohexene (C6H10) formation on TTAB-stabilized cubic and cuboctahedral Pt nanoparticles (NPs) for 1.3×104 Pa C6H6, 1.3×105 Pa H2, and 8.5×105 Pa Ar, and (b) corresponding Arrhenius plots; Dotted lines are drawn as visual aides[62]
图11
(a) 在1.3×104 Pa C6H6, 1.3×105 Pa H2和8.5×105Pa Ar的条件下, 十四烷基三甲基溴化铵(TTAB)稳定的Pt立方相和Pt立方八面相对环己烷和环己烯的转换率; (b)相应的阿伦尼乌斯图[62]
Figure11.
(a) Turnover rates of cyclohexane (C6H12) and cyclohexene (C6H10) formation on TTAB-stabilized cubic and cuboctahedral Pt nanoparticles (NPs) for 1.3×104 Pa C6H6, 1.3×105 Pa H2, and 8.5×105 Pa Ar, and (b) corresponding Arrhenius plots; Dotted lines are drawn as visual aides[62]
另外通过改变载体的形貌也可以对催化性能产生影响。Shi等[64]研究了金纳米粒子负载在不同相的CeO2(棒状,立方块,多面体)上对水煤气转化反应的影响,相比其它形状的CeO2,由(110)和(100)晶面的棒状CeO2的反应活性最高。这是由于棒状CeO2纳米颗粒(110)晶面上氧空位生成使其反应活化能明显低于其它形貌的CeO2。
2.3 组成因素
对于金属纳米复合材料,特别是双金属催化剂或者金属合金,由于引入其他金属组分可以调节和改变催化剂的形貌和电子特性,从而提高催化剂的活性和选择性。在金属复合催化剂中,由于不同组分的结构失配,导致晶格膨胀或收缩,改变金属d带中心和宽度,从而影响与吸附物的结合能[65-66],改变反应速率和反应途径。
Yu等[67]以乙酰丙酮镍和乙酰丙酮铜为原料制备出16 nm单分散的不同铜镍比的合金并将铜镍合金纳米粒子分散在石墨烯上(G-CuNi NPs)。通过TEM可以看出制备的Cu36Ni64 NPs的平均大小为(16.0±1.0) nm。根据高分辨透射电镜可以看出Cu36Ni64 NPs的晶格间距为0.205 nm,小于铜的0.208 nm,大于镍的0.203 nm (图 13B),证明铜镍合金是以固溶体的形式存在。根据高角度环形暗场(HAADF)扫描透射电子显微镜和元素分布可以看出铜镍在纳米粒子上是分布均匀分布(图 13C)。该催化剂通过丁胺和乙醇洗涤活化,研究了不同比例铜镍催化剂对氨硼烷醇解产氢的影响。铜镍合金的催化性能明显优于铜和镍,其中G-Cu36Ni64 NPs显示出最优的催化活性。这种催化性能的提升源于合金效应。铜镍一旦形成合金,d带中心位置移动到了铜(-2.67 eV)和镍(-1.29 eV)之间。d带中心位置的改变可以优化催化物种与催化剂表面的结合能,从而影响催化性能。
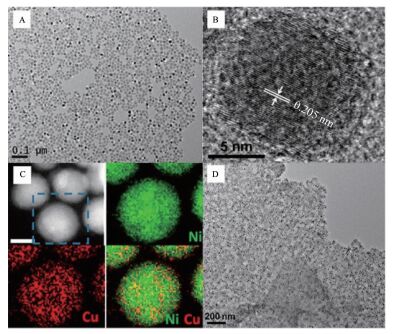 图13
(A) Cu36Ni64 NPs的透射电镜图; (B)Cu36Ni64 NPs高分辨透射电镜图; (C)高角度环形暗场扫描透射电子显微镜图和元素分布图Ni(绿色)铜(红色); (D)氨硼烷处理的G-Cu36Ni64 NPs的透射电镜图[67]
Figure13.
(A) TEM image of the as-prepared Cu36Ni64 NPs; (B) HRTEM image of a single Cu36Ni64 NP; (C) HAADF-STEM image of the Cu36Ni64 NPs (scale bar: 10 nm) and STEM-EELS elemental mapping of a selected NP to show Ni (green) and Cu (red) distribution within the NP; (D) TEM image of the ammonia borane-treated G-Cu36Ni64 NPs[67]
图13
(A) Cu36Ni64 NPs的透射电镜图; (B)Cu36Ni64 NPs高分辨透射电镜图; (C)高角度环形暗场扫描透射电子显微镜图和元素分布图Ni(绿色)铜(红色); (D)氨硼烷处理的G-Cu36Ni64 NPs的透射电镜图[67]
Figure13.
(A) TEM image of the as-prepared Cu36Ni64 NPs; (B) HRTEM image of a single Cu36Ni64 NP; (C) HAADF-STEM image of the Cu36Ni64 NPs (scale bar: 10 nm) and STEM-EELS elemental mapping of a selected NP to show Ni (green) and Cu (red) distribution within the NP; (D) TEM image of the ammonia borane-treated G-Cu36Ni64 NPs[67]
不同组分之间的电子效应也可以改变表面金属的电子状态。Wang等[68]合成了以石墨烯为载体的Au-Pd催化剂,该催化剂对甲醇选择性氧化合成甲酸甲酯具有很好的活性,Au和Pd之间的电子转移能够提高催化剂的催化活性。其中,Au2.0-Pd1.0/石墨烯催化剂中Au和Pd电子结合能均变低证明了Au和Pd之间存在着电子转移。
2.4 配体和载体的影响
在金属纳米粒子的合成过程中,由于纳米粒子的高活性因此很容易被氧化,导致催化活性显著下降。在金属纳米粒子的合成过程中加入有机配体,不仅可以提高金属纳米粒子的稳定性,同样由于配体与金属之间的空间效应也可以影响其反应活性。Wu等[69]合成了有机胺包覆的PtCo的纳米晶。该工作研究了不同链长的有机胺对催化性能的影响。从图 14a可以看出,随着链的减小,肉桂醇的选择性逐渐降低。PtCo的纳米晶被C18NH2,OAm,C16NH2和C12NH2包覆时,在肉桂醛的转化率为100%时,肉桂醇的选择性均能达到90%以上。相比之下,C8NH2包覆的PtCo的纳米晶对肉桂醇的选择性为78%。当用C4NH2包覆时,肉桂醇的选择性仅为23%。以上实验数据表明该催化剂表面存在着长链的有机胺化合物,由于立体位阻效应,不利于C=C的吸附,提高了C=O的选择性还原。短链有机胺包覆的PtCo的纳米晶有助于肉桂醛在催化剂表面的分布,提高了加氢能力(图 14b),肉桂醇能够进一步被还原,降低了肉桂醇的选择性。这项研究显示了PtCo纳米晶的催化活性依赖于表面有机胺链的长短。短链有机胺有助于提高反应活性,长链有机胺有利于提高选择性。
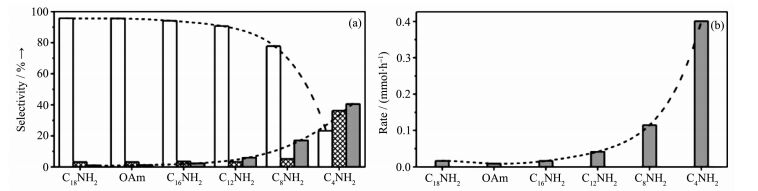 图14
(a) 不同有机胺包覆的Pt3Co催化剂在肉桂醛接近100%转化时肉桂醇(白色), 氢化肉桂醛(阴影), 氢化肉桂醇(灰色)的选择性直方图; (b)在肉桂醛完全转化时, 不同有机胺包覆的Pt3Co催化剂的氢化肉桂醇产生速率[69]
Figure14.
Histograms of (a) Selectivity for COL (white), HCAL (crosshatch), and HCOL (gray) at nearly 100% conversion in the hydrogenation of CAL with different amine-capped Pt3Co catalysts; (b) Rates of HCOL production after complete conversion of CAL with different amine-capped Pt3Co nanocatalysts[69]
图14
(a) 不同有机胺包覆的Pt3Co催化剂在肉桂醛接近100%转化时肉桂醇(白色), 氢化肉桂醛(阴影), 氢化肉桂醇(灰色)的选择性直方图; (b)在肉桂醛完全转化时, 不同有机胺包覆的Pt3Co催化剂的氢化肉桂醇产生速率[69]
Figure14.
Histograms of (a) Selectivity for COL (white), HCAL (crosshatch), and HCOL (gray) at nearly 100% conversion in the hydrogenation of CAL with different amine-capped Pt3Co catalysts; (b) Rates of HCOL production after complete conversion of CAL with different amine-capped Pt3Co nanocatalysts[69]
催化剂载体对催化剂催化活性有着十分重要的影响。除了提高金属纳米粒子分散性。更重要的是,载体与金属之间的相互作用可以影响催化反应的活性和选择性。为了更好的研究金属与载体的相互作用对催化反应的影响,Griffin等[70]以Pt/TiO2和Pt/SiO2为催化剂,通过气相条件下还原巴豆醛和原位和频光谱测量技术,研究不同载体对还原巴豆醇的影响。Pt/TiO2的催化活性是Pt/SiO2的4倍,对巴豆醇的选择性为50%以上。为了更好的理解TiO2对于C=O的还原的促进作用,利用原位和频光谱测量技术检测反应过程中的中间产物。Pt/SiO2显示了2个波谱峰2 860和2 915 cm-1,其中2 860 cm-1归因于醛类化合物的CH的伸缩振动,2 915 cm-1归属于CH3的伸缩振动(图 15A)。以上的吸收峰证明了以Pt/SiO2为催化剂时,产生了丁烯醛,对于加氢还原巴豆醛没有催化活性。对于Pt/TiO2,出现了2 880,2 930,2 965 cm-1。其中2 880,2 930 cm-1分别归属于CH3的对称伸缩振动和不对称伸缩振动,2 965 cm-1归属于CH2键在C-O键上的非对称伸缩振动(图 15B)。测试结果说明在TiO2存在着巴豆醛基-氧的中间体。这是由于Pt与TiO2的相互作用,H由Pt溢流到TiO2上,使TiO2被还原为TiO2-x,金属性增强,氧空位增加,TiO2与巴豆醛键合形成巴豆醛基-氧的中间体。再通过H由Pt溢流到TiO2上实现对C=O的还原。
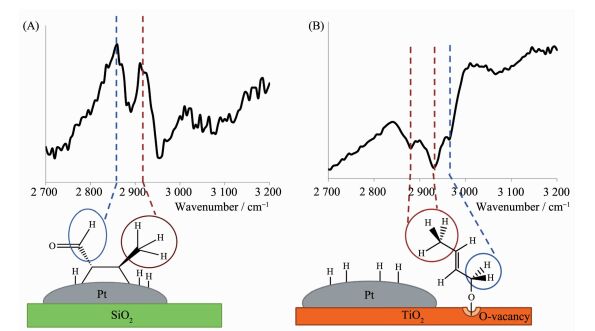 图15
和频光谱图(A) Pt/SiO2和(B)Pt/TiO2在反应温度120 ℃, 1.33×102 Pa巴豆醛, 1.33×104 Pa氢气和8.92×104 Pa氩气下; 文中提到的这些光谱图是相应的表面物种[70]
Figure15.
SFG spectra for (A) Pt/SiO2 and (B) Pt/TiO2 under reaction conditions of 1.33×102 Pa crotonaldehyde, 1.33×104 Pa hydrogen, and 8.92×104 Pa argon at a catalyst temperature of 120 ℃; Assignment of these spectra, explained in the text, yields the accompanying surface intermediates[70]
图15
和频光谱图(A) Pt/SiO2和(B)Pt/TiO2在反应温度120 ℃, 1.33×102 Pa巴豆醛, 1.33×104 Pa氢气和8.92×104 Pa氩气下; 文中提到的这些光谱图是相应的表面物种[70]
Figure15.
SFG spectra for (A) Pt/SiO2 and (B) Pt/TiO2 under reaction conditions of 1.33×102 Pa crotonaldehyde, 1.33×104 Pa hydrogen, and 8.92×104 Pa argon at a catalyst temperature of 120 ℃; Assignment of these spectra, explained in the text, yields the accompanying surface intermediates[70]
3 金属纳米复合材料在还原不饱和化学键中的应用
不饱和化学键存在于大量有机化合物中,还原有机化合物中的不饱和化学键制备高附加值的化学品广泛应用于工农业生产中[71]。金属纳米复合材料催化剂在这方面显示了优异的催化性能。我们以典型不饱和化学键为例,包括芳香族硝基化合物(N=O),腈类化合物(N≡C),α,β-不饱和醛(C=O),讨论金属纳米复合材料在还原不饱和化学键中的应用情况。
3.1 芳香族硝基化合物的还原
芳香族硝基化合物是一种高毒性的有机化合物,对环境,人类和动植物具有潜在的危害性[72-73]。其中将芳香族硝基化合物还原为无毒的胺基类化合物是一个重要的解决途径,另外还原得到的胺基类化合物可以作为中间体广泛应用于精细化工、生物医药中。但是,由于芳香族硝基化合物的化学惰性,仅在还原剂的存在下很难发生反应,因此选取合适的催化剂是必要的。同时,还原芳香族硝基化合物可以作为模型反应,应用于金属纳米复合催化剂的性能研究和催化反应机理讨论。目前,各种金属纳米复合催化剂在还原芳香族硝基化合物到胺基类化合物中得到了广泛的应用(见表 1)。
 表 1
各种金属纳米复合催化剂应用于还原芳香族硝基化合物到胺基类化合物
Table 1.
Summary of the reduction of aromatic nitro-compounds catalyzed by metal catalysts
表 1
各种金属纳米复合催化剂应用于还原芳香族硝基化合物到胺基类化合物
Table 1.
Summary of the reduction of aromatic nitro-compounds catalyzed by metal catalysts

一般情况下,在还原芳香族硝基化合物到胺基类化合物的过程中,贵金属催化剂得到了广泛的使用。很多研究小组致力于利用不同方法将贵金属纳米粒子稳定和分散在不同的载体上,以提高其对还原芳香族硝基化合物的反应活性。Mohanta等[77]采用CTAB将金纳米棒封装在不同厚度(10.1,12.1和14.1 nm)的多孔二氧化硅内(Au@SiO2)(图 16)。通过透射电镜可以看出用多孔二氧化硅封装后的金纳米棒的平均直径是(15±2) nm。一层薄薄的多孔二氧化硅覆盖在整个金纳米棒的表面,二氧化硅表面的粗糙度也表明孔隙的存在。3个不同厚度二氧化硅(10.1,12.1和14.1 nm)封装的金纳米棒被命名为Au NR@SiO2-10, Au NR@SiO2-12和Au NR@SiO2-14。EDX也证明多孔二氧化硅覆盖在整个金纳米棒的表面(图 16D)。封装后的金纳米粒子对还原对硝基苯酚的反应活性(k=2.92×10-1 mol·L-1·min-1)是未封装(k=3.74×10-2 mol·L-1·min-1)的8倍(图 16E)。催化剂的反应活性同样也与多孔二氧化硅的厚度有关,随着多孔二氧化硅的厚度降低,反应速率逐渐增加。多孔二氧化硅层覆盖在金纳米棒的表面不仅稳定了金纳米粒子,也提供一个通道易于反应分子通过金属界面,促进反应的进行。该工作为提高金纳米粒子的催化活性提供了一个新的思路。另外,Fu等[74]通过碳材料的高温碳化得到金纳米粒子插层在有序介孔碳纳米球的框架中。碳材料既可以作为还原剂获得金纳米粒子,同样也起到了稳定纳米粒子的作用。通过有关表征可以得出大小为2.8 nm的金纳米粒子被插嵌到形貌均一的介孔碳球(90 nm)框架中。该催化剂在氢气的作用下对还原芳香族硝基化合物到胺基类化合物显示出高的催化活性和选择性。催化剂高的活性和选择性依赖于高温还原过程中形貌的改变以及低价态的金物种的出现。除了金纳米粒子催化剂,其他贵金属纳米粒子(Pd,Ag,Pt等)也应用于还原芳香族硝基化合物[79, 86, 91]。Nandi等[82]利用化学还原法将钯纳米颗粒分散在氮化碳上,应用于还原芳香族硝基化合物。以联氨为还原剂在优化的反应条件下,一系列的芳香族硝基化合物可以转化为相应的胺类化合物,产率从80%到99%,显示出了高活性和高选择性。该催化剂可以多次循环使用而不影响催化性能,也可以长时间保存并保持较高的催化效率。
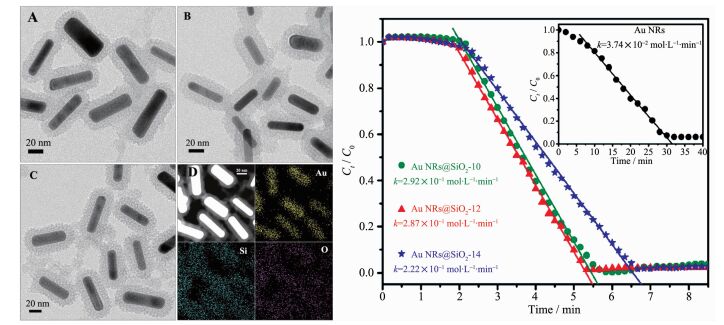 图16
(A~C)不同厚度的介孔二氧化硅包覆的金纳米棒的高分辨透射电镜: (A) Au NRs@SiO2-10, (B) Au NRs@SiO2-12, (C) Au NRs@SiO2-14; (D) Au NRs@SiO2-10的元素分布; (E)不同厚度的介孔二氧化硅包覆的金纳米棒催化对硝基苯酚的动力学常数(Au NRs@SiO2-10、AuNRs@SiO2-12和Au NRs@SiO2-14), 插图为CTAB-封装的Au NRs的动力学常数[77]
Figure16.
(A~C) HRTEM images of gold nanorods after coating with a layer of porous silica of varying shell thickness:(A) Au NRs@SiO2-10, (B) Au NRs@SiO2-12, (C) Au NRs@SiO2-14; (D) EDX elemental mapping of Au NRs@SiO2-10; (E) Kinetic data for the catalysis of 4-nitrophenol using various silica-coated gold nanorods (Au NRs@SiO2-10, AuNRs@SiO2-12, and Au NRs@SiO2-14); Inset shows the linear fitting of kinetic data with CTAB-capped Au NRs[77]
图16
(A~C)不同厚度的介孔二氧化硅包覆的金纳米棒的高分辨透射电镜: (A) Au NRs@SiO2-10, (B) Au NRs@SiO2-12, (C) Au NRs@SiO2-14; (D) Au NRs@SiO2-10的元素分布; (E)不同厚度的介孔二氧化硅包覆的金纳米棒催化对硝基苯酚的动力学常数(Au NRs@SiO2-10、AuNRs@SiO2-12和Au NRs@SiO2-14), 插图为CTAB-封装的Au NRs的动力学常数[77]
Figure16.
(A~C) HRTEM images of gold nanorods after coating with a layer of porous silica of varying shell thickness:(A) Au NRs@SiO2-10, (B) Au NRs@SiO2-12, (C) Au NRs@SiO2-14; (D) EDX elemental mapping of Au NRs@SiO2-10; (E) Kinetic data for the catalysis of 4-nitrophenol using various silica-coated gold nanorods (Au NRs@SiO2-10, AuNRs@SiO2-12, and Au NRs@SiO2-14); Inset shows the linear fitting of kinetic data with CTAB-capped Au NRs[77]
基于贵金属的价格昂贵和稀缺性,设计高反应活性,高稳定的非贵金属催化剂应用于芳族硝基化合物的还原是十分有必要的。Yang等[93]引入简单的电位移反应,成功的将铜纳米粒子负载在硅纳米线阵列(Si NWAs)上。硅纳米线阵列能够很好的稳定和分散铜纳米粒子,使其具有可控的尺寸和表面覆盖率。负载在硅纳米线阵列的铜纳米粒子在还原对硝基苯酚到对氨基苯酚的反应中展现出高的反应活性和稳定性,反应5次以后,对硝基苯酚的转化率还能高达95%。更重要的是,铜纳米粒子能够被移除和替代,硅纳米线阵列不被破坏且能重复利用。除了铜基催化剂,镍基催化剂应用于还原硝基化合物也有相关报道。Jiang等[47]利用原子沉积的方法,将镍纳米粒子高度分散到多孔的二氧化硅上(Ni/SiO2)。以联氨为还原剂,Ni/SiO2为催化剂应用于芳香族硝基化合物的还原,表现出较高的反应活性和选择性。
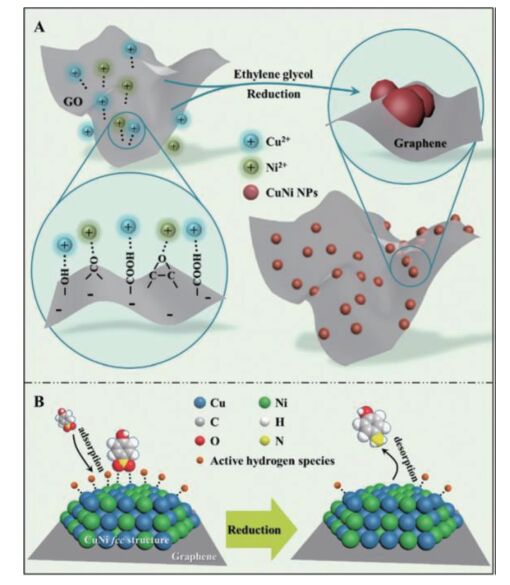
不同于单一金属催化剂,由于金属合金中不同元素之间的协同效应,金属合金类催化剂的催化效果明显优于单一金属的,因此金属合金类催化体系也被应用于芳香族硝基化合物的还原[82, 90]。Fang等[92]利用静电吸附和原位还原制备了CuNi合金纳米粒子负载在石墨烯上形成二维结构纳米复合催化剂。图 17A显示了该复合催化剂的形成机理。在静电吸附的作用下,铜和镍的金属离子被吸附在石墨烯表面的-OH,-OOH和其他含氧基团。然后通过加热乙二醇原位还原石墨烯和铜镍金属离子,最终实现超小的CuNi纳米粒子(2 nm)均匀地锚定在石墨烯上。石墨烯/ CuNi纳米复合材不仅实现了结构的突破,而且在活性和稳定性方面也展现了优异的性能。通过改变铜镍合金的比例,石墨烯/Cu61Ni39的反应速率常数可高达0.136 85 s-1,各类芳香族硝基化合物的选择性高达99%。作者也提出了铜镍合金催化还原芳香族硝基化合物的反应机理(图 17B)。首先在铜镍纳米粒子的催化作用下,硼氢化物产生活化的氢物种并与铜镍合金结合。同时,吸附的对硝基苯酚离子被这些活化的氢物种还原形成对氨基苯酚。由于对氨基苯酚的吸附能力明显小于对硝基苯酚,一旦在催化剂表面形成很快就能解离。考虑到吸脱附的速率很快,整个反应速率被硝基的还原控制。因此,还原芳香族硝基化合物被认为是一级反应。
3.2 腈类化合物的还原
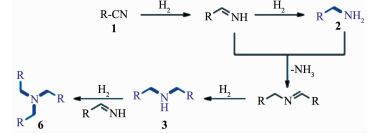
胺类化合物在精细化工、制药、材料科学和生物技术得到广泛的应用, 其中还原腈类化合物是一步法合成多元胺的有效途径[94-95]。然而, 由于在还原腈类化合物的过程中存在着胺类混合物比如二级胺、亚甚至叔胺等(图 18)。选择性还原腈类化合物合成胺类化合物具有一定的挑战性。目前,均相和异相的过渡金属配合物是选择性还原腈类化合物的主要催化剂[96]。随着纳米科学的发展,金属催化剂特别是金属纳米复合催化剂可以替代过渡金属配合物来控制这个反应的选择性。
Wang等[97]报道了Pd@g-C3N4催化剂在选择性还原腈类化合物中展现了较高的转化率和选择性(高到99%)。在最优的反应条件下,芳香族腈类化合物可以转化为三级胺类化合物。他们还遴选了其他钯基催化剂比如Pd/TiO2,Pd/CeO2和Pd/Al2O3,以上催化剂同样可以实现将腈基化合物转化为三级胺类化合物。该工作的主要优点是使用常压氢气,无反应溶剂和良好的循环性能。
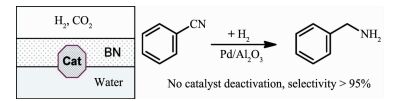
Arai等[98]在二氧化碳和水的存在下,使用Pd/Al2O3可以高效的转化苯腈到苯胺(图 19)。由于水的存在苯胺进入水相,避免与中间产物亚胺反应生成二级胺。另外,二氧化碳可以与苯胺结合,从而提高苯胺的选择性。除了钯基催化剂,Lu等[99]以铂纳米线为催化剂,选择性还原腈类化合物到合成二级胺。这种方法可以在存在其他胺或者不存在的情况下,合成不对称和对称的仲胺。
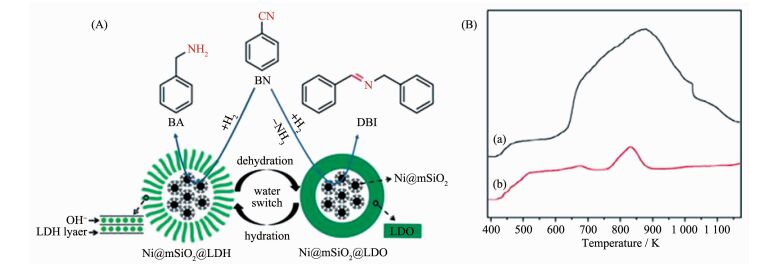
一些含量丰富,价格低廉的非贵金属催化剂也被应用于还原腈类化合物到胺类化合物催化反应中。Marella等[100]采用浸渍的方法制备出不同负载量的Cu-MgO2催化剂。随着铜的负载量的增加,苯胺的转化率逐渐增加,当负载量达到12%时,显示出最高的催化活性(98%的苯腈转化率,70%的苯胺选择性)。当负载量超高12%,催化性降低,这是由于大颗粒的铜粒子形成。通过优化反应条件,苯腈完全转化,苯胺的选择性能够达到99%。Cao等[101]以类水滑石前驱体,通过水解和煅烧的两种方式制备出了双氢氧化物包覆的镍基核壳结构的纳米粒子(Ni@mSiO2@LDH)和双氧化物包覆的镍基核壳结构的纳米粒子(Ni@mSiO2@LDO)。Ni@mSiO2@LDH和Ni@mSiO2@LDO分别将苯腈选择性还原为苯胺(BA)和N-苄亚甲基苄胺(DBI)(图 20A),选择性分别为76.1%和92.3%。这种性能的差异是由于水解过程和煅烧过程导致外层类水滑石结构和酸碱性的改变。通过图 20B可以看出Ni@mSiO2@LDO的脱附峰明显大于Ni@mSiO2@LDH的, 表明Ni@mSiO2@ LDO的酸性高于Ni@mSiO2@LDH。在高酸性条件下,有利于反应过程中产生的中间体亚胺和苯胺结合生成N-苄亚甲基苄胺(DBI)。相反的是,在水解过程中,OH-进入Ni@mSiO2@LDH的夹层中降低了催化剂的酸性,这种改变提高了苯胺的选择性。这种方法可以有效的设计催化剂应用于提高其他催化反应的选择性。
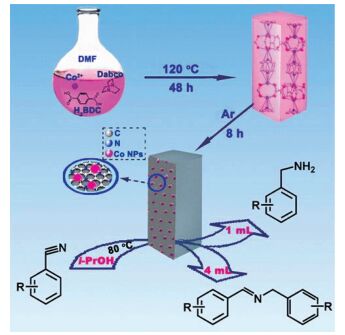
在非氢气的条件下,利用催化转移加氢的方法还原腈类化合物合成一级胺或者亚胺是一个重要和替代的策略。发展高活性双功能的催化剂体系进行选择性反应仍然是一个巨大的挑战。Long等[102]制备了氮掺杂的Co@NC催化体系应用于催化转移加氢还原腈类化合物(图 21)。通过在惰性气体条件下,高温分解氮掺杂的Co-MOF获得Co@NC。氮的引入得到了高质量的氮掺杂的石墨烯,使催化剂具有较高的碱性位点。钴离子被还原为钴纳米粒子并均匀分布在氮掺杂的石墨烯上。在不添加碱促进剂的条件下,以异丙醇为氢源和溶剂,改变异丙醇的体积可以选择性获得伯胺和亚胺。氮的引入起到与碱促进剂相似的作用,不仅可以阻止聚胺的形成,同样也可以防止反应产物在催化剂表面的聚集,从而提高催化转移加氢还原腈类化合物的效率。这个工作为在无碱促进剂和分子氢的条件下,为催化转移加氢还原腈类化合物到伯胺和亚胺提供了一个新的策略。
3.3 α,β-不饱和醛的选择性还原
α,β-不饱和醛是重要的化工原料及中间体,其中将C=O双键选择性还原为α,β-不饱和醇可以广泛应用于调味品,香料和药物合成[103-104]。选择性还原α,β-不饱和醛到α,β-不饱和醇存在着一定的挑战。在热力学和动力学上C=C比C=O更容易还原[105-107]。另外,当α,β-不饱和醛在金属表面吸附时,存在着C=C和C=O的竞争关系,导致选择性还原C=O的能力下降[108]。为了提高金属催化剂对选择性还原C=O的催化活性,很多研究者致力于对金属催化剂进行改性,设计合成高活性和高选择性的金属纳米复合催化剂应用于选择性还原α,β-不饱和醛到α,β-不饱和醇。
3.3.1 引入助催化剂
Tamura等[109]通过将铁的金属盐(FeCl3,Fe(NO3)3, FeCl2,FeSO4)和Ir/MgO混合应用于α,β-不饱和醛(酮)的选择性还原(图 22)。当以苄叉丙酮为反应物时,以Ir/MgO为催化剂,苄叉丙酮的转化率仅为15%,不饱和醇的选择性为13%,当在该催化体系下加入铁的金属盐,苄叉丙酮的转化率和不饱和醇的选择性增加明显。其中当加入Fe(NO3)3后,苄叉丙酮的转化率和不饱和醇的选择性分别是58%和85%。根据动力学研究和催化剂的表征,活性位点被认为是金属钇,Ir4+和Fe2+物种在氧化镁载体上形成的界面和通过活化的氢物种(H-和H+)形成的阴-阳离子活性位点Fe2+-O2-,从而提高反应活性和选择性。Yang等[110]在纳米镍催化剂中加入氧化锌应用于液相还原柠檬醛到香茅醇。实验结果发现氧化锌的引入提高了香茅醇的选择性。氧化锌和金属镍的相互作用改变了镍活性位点的电子结构,阻碍了C=C的吸附。氧化锌作为一种路易斯酸物种提高了对C=O双键的吸附能力,从而获得香茅醇高的选择性(>92%)。
3.3.2 载体的引入
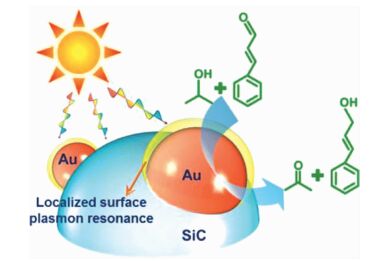
在金属纳米复合材料催化剂体系中,载体与金属之间的相互作用可以影响催化反应的活性和选择性。Hao等[103]在可见光的驱动下,以异丙醇为氢源,Au/SiC可以高效的选择性光还原肉桂醛到肉桂醇。Au/SiC优异的催化活性依赖于Au纳米粒子的表面等离子体效应和Au/SiC之间的电子转移(图 23)。在可见光的驱动下,异丙醇被氧化为丙酮,产生活性氢。同时,在表面等离子体活化的金纳米粒子的作用下,优先吸附的端位的醛基被加氢还原为醇。
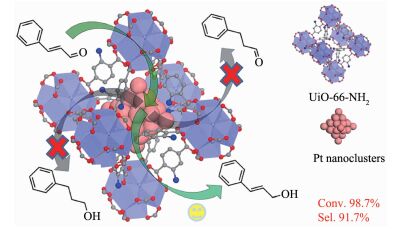
载体的结构不仅影响催化体系的电子效应也可以在立体空间上影响C=C和C=O吸附取向模式。Huang等[111]合成了氨基修饰的MOFs(UiO-66-NH2),由于氨基的存在,金属离子极易进入MOFs的孔道中,使得Pt纳米粒子保留在MOFs孔道中。根据透射电镜的测试结果,0.97%(w/w)的Pt纳米粒子的粒径为1.2 nm左右,小于UiO-66-NH2的孔径。更重要的是,将负载量由0.97%(w/w)提高到10.7%(w/w),Pt纳米粒子的粒径大小仍基本保持不变,在MOFs孔内增加负载量并没有影响Pt纳米粒子的粒径大小。为了证明Pt@UiO-66-NH2的立体空间选择性,作者将其应用到催化还原肉桂醛的反应中,由于Pt纳米粒子处在MOFs孔道内部,而肉桂醛必须要进入MOFs孔道才能与Pt纳米粒子接触,因此,端基上的醛基可以被加氢还原,而中间的C=C双键很难被加氢还原,醛基加氢的选择性为91.7%(图 24)。载体除了能增加C=O的吸附能力,还能抑制催化剂与C=C键吸附能力。
3.3.3 引入有机配体
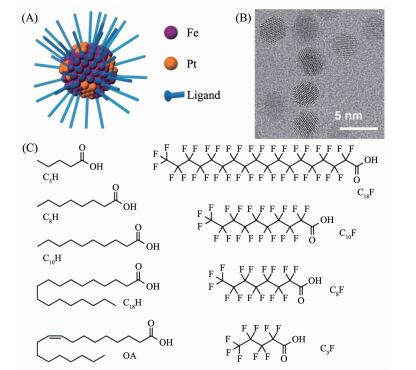
在金属纳米粒子的合成过程中加入有机配体,不仅可以提高金属纳米粒子的稳定性,更重要的是,配体与金属之间的相互作用和空间效应可以影响其反应活性和选择性。Khanh等[112]通过加入各种羧酸盐有机配体来稳定FePt纳米粒子。这些有机配体可以选择性与Fe键合(图 25A)。其中通过碳氢化合物类有机配体稳定的FePt纳米粒子对于还原肉桂醛显示出了较低的催化活性和选择性,而全氟类有机配体稳定的FePt纳米粒子可以明显提高选择性还原肉桂醛到肉桂醇的催化活性。催化剂表面的有机配体,造成了Pt活性位点被孤立,改变了活性位点附近的空间构型,易于与端位的C=O结合,提高肉桂醇的选择性。相比于碳氢类化合物,全氟类有机配体在结构上更加刚性,几何空间效应更加明显,利于选择性还原肉桂醛到肉桂醇。另外,有机配体选择性与Fe键合,随着Fe含量的增加,表面配体密度增加,因此高的Fe含量更有利于选择性还原C=O。
4 总结与展望
在本文中,我们综述了金属纳米复合催化剂的最新进展。由于金属纳米复合催化剂结合了多种组分的性能,在催化反应中得到广泛应用,并显示出了优异的反应活性和选择性。首先,我们主要总结了金属纳米复合催化剂的合成方法。随着纳米材料合成方法的发展,具有特定尺寸,形貌以及催化性能的金属纳米复合材料可以通过各种合成方法得到。为了更好的优化催化剂的结构与催化活性的关系,并理解催化反应机理,我们讨论了金属纳米复合催化剂一些重要因素(如粒子粒径,形貌,组成,载体或配体)对催化活性和选择性的影响。最后,本文总结了近几年来金属纳米复合催化剂在还原不饱和化学键的应用。其中主要列举了金属纳米复合催化剂在催化还原芳香族硝基化合物,腈类化合物以及选择性催化还原α,β-不饱和醛的研究进展。
目前,金属纳米复合催化剂仍然存在一些需要解决和值得研究的问题:(1)在金属纳米复合催化剂制备过程中,充分认知和理解金属纳米材料与其他材料之间的结构关系,对于设计高效的金属纳米复合催化剂是十分重要的;(2)现阶段金属纳米复合催化剂仅限于一些典型的模型反应和实验阶段,鲜有大规模的应用的报道,限制了金属纳米复合催化剂在工业化的应用。如何解决宏观制备过程中的技术瓶颈是目前急待解决的问题;(3)在金属纳米复合催化剂中,贵金属被大量应用,很大程度上提高了化学反应的成本。为了提高贵金属催化剂原子的利用率和反应活性,制备单原子贵金属催化剂是一种的新趋势;(4)在金属纳米催化剂中引入其他组分形成金属纳米复合催化剂,比如形成多元合金,制备负载性金属催化剂,引入配体或者助催化剂可以提高催化性能,往往归因与不同组分之间的相互作用。在实际的反应过程中很难准确理解催化体系中各种组分对催化反应的影响。在反应过程中,引入一些原位表征技术可以在分子层面上直接获取催化行为的相关信息,可以更好的研究金属纳米复合催化剂结构与催化性能的关系。
发展高效且应用广泛的金属纳米复合材料仍需要我们深入研究。通过深入理解金属纳米复合材料中各组分之间的结合机理和相互作用,可以拓宽金属纳米复合材料的应用范围,将使其在催化反应和其他领域拥有更加广阔的前景。
-
-
[1]
Zhou K B, Li Y D. Angew. Chem. Int. Ed., 2012, 51(3):602-613 doi: 10.1002/anie.201102619
-
[2]
Xu Y, Chen L, Zhang Q, et al. Nanoscale, 2015, 7(24):10559-10583 doi: 10.1039/C5NR02216A
-
[3]
Dai Y H, Wang Y, Yang Y H. Small, 2015, 11(3):268-289 doi: 10.1002/smll.v11.3
-
[4]
Li H, Liao J Y, Du Y H, et al. Chem. Commun., 2013, 49(17): 1768-1770 doi: 10.1039/c3cc39237f
-
[5]
Sahoo G P, Bhui D K, Misra A, et al. J. Mol. Liq., 2014, 198: 215-222 doi: 10.1016/j.molliq.2014.06.032
-
[6]
Hu H W, Xin J H, Hu H, et al. J. Mater. Chem. A, 2015, 3 (21):11157-11182 doi: 10.1039/C5TA00753D
-
[7]
Sharma N, Ojha H, Sharma R K, et al. RSC Adv., 2015, 5(66): 53381-53403 doi: 10.1039/C5RA06778B
-
[8]
Sun Y, Zhang F, Xu L, et al. J. Mater. Chem. A, 2014, 2(43): 18583-18592 doi: 10.1039/C4TA03689A
-
[9]
Zhang P, Shao C, Zhang Z, et al. Nanoscale, 2011, 3(8):3357-3363 doi: 10.1039/c1nr10405e
-
[10]
Villa A, Wang D, Dimitratos N, et al. Catal. Today, 2010, 15: 8-15
-
[11]
Ma G, Yan X, Li Y, et al. J. Am. Chem. Soc., 2010, 132 (28):9596-9597 doi: 10.1021/ja1027524
-
[12]
Zhang Q, Lee I, Ge J P, et al. Adv. Funct. Mater., 2010, 20 (14):2201-2214 doi: 10.1002/adfm.v20:14
-
[13]
Li Z W, Li M, Bian Z F, et al. Appl. Catal., B, 2016, 188: 324-341 doi: 10.1016/j.apcatb.2016.01.067
-
[14]
Ferrando R, Jellinek J, Johnston R L. Chem. Rev., 2008, 108 (3):845-910 doi: 10.1021/cr040090g
-
[15]
Liu P L, Gu X J, Kang K, et al. ACS Appl. Mater. Interfaces, 2017, 9(12):10759-10767 doi: 10.1021/acsami.7b01161
-
[16]
Akbayraka S, Tonbulb Y zkara S. Appl. Catal., B, 2017, 206:384-392 doi: 10.1016/j.apcatb.2017.01.063
-
[17]
Gu D, Tseng J C, Weidenthaler C, et al. J. Am. Chem. Soc., 2016, 138(30):9572-9580 doi: 10.1021/jacs.6b04251
-
[18]
Nie D, Xu C J, Chen H Y, et al. Mater. Lett., 2014, 13:1306-1309
-
[19]
Truong P L, Ma X Y, Sim S J. Nanoscale, 2014, 6(4):2307-2315 doi: 10.1039/c3nr05211g
-
[20]
Zaina N M, Stapleya A G F, Sham G. Carbohydr. Polym., 2014, 112:195-202 doi: 10.1016/j.carbpol.2014.05.081
-
[21]
Wu K L, Wei X W, Zhou X M, et al. J. Phys. Chem. C, 2011, 115(33):16268-16274 doi: 10.1021/jp201660w
-
[22]
Sun S, Zhang G, Geng D, et al. Angew. Chem. Int. Ed., 2011, 50(2):422-426 doi: 10.1002/anie.201004631
-
[23]
Gupta V K, Atar N, Uzun L, et al. Water Res., 2014, 48:210-217 doi: 10.1016/j.watres.2013.09.027
-
[24]
Wang M L, Wang L B, Li H L, et al. J. Am. Chem. Soc., 2015, 137(44):14027-14030 doi: 10.1021/jacs.5b08289
-
[25]
Zhang Y L, Li L P, Li G S, et al. Chem. Eur. J., 2016, 22 (40):14196-14204 doi: 10.1002/chem.201601681
-
[26]
Lü H F, Xi Z, Chen Z Z, et al. J. Am. Chem. Soc., 2015, 137(18):5859-5862 doi: 10.1021/jacs.5b01100
-
[27]
Huang S S, He Q Q, Zai J T, et al. Chem. Commun., 2015, 51 (43):8950-8953 doi: 10.1039/C5CC02584B
-
[28]
Nam K M, Shim J H, Ki H, et al. Angew. Chem. Int. Ed., 2008, 7(49):9504-9508
-
[29]
Chaubey G S, Barcena C, Poudyal N, et al. J. Am. Chem. Soc., 2007, 129(23):7214-7215 doi: 10.1021/ja0708969
-
[30]
Yu Y S, Yang W W, Sun X L, et al. Nano Lett., 2014, 14(5): 2778-2782 doi: 10.1021/nl500776e
-
[31]
Niu G D, Zhou M, Yang X, et al. Nano Lett., 2016, 16(6): 3850-3857 doi: 10.1021/acs.nanolett.6b01340
-
[32]
Zeng D Q, Gong P Y, Chen Y Z, et al. Nanoscale, 2016, 8 (22):11602-11610 doi: 10.1039/C6NR02055K
-
[33]
Buceta D, Tojo C, Vukmirovic M B, et al. Langmuir, 2015, 31(27):7435-7439 doi: 10.1021/acs.langmuir.5b01455
-
[34]
Montes V, Boutonnet M, Jaras S, et al. Catal. Today, 2014, 223:66-75 doi: 10.1016/j.cattod.2013.11.053
-
[35]
MartΙńez-Rodrģuez R A, Vidal-Iglesias F J, Solla-Gulloń J, et al. J. Am. Chem. Soc., 2014, 136(4):1280-1283 doi: 10.1021/ja411939d
-
[36]
MartΙńez-Rodrģuez R A, Vidal-Iglesias F J, Solla-Gulloń J, et al. ChemPhysChem, 2014, 15(10):1997-2001 doi: 10.1002/cphc.v15.10
-
[37]
李峰, 梁晋榕, 任泉明, 等.能源化工, 2016, 37(4):44-48LI Feng, LIANG Jin-Rong, REN Quan-Ming, et al. Energy Chemical Industry, 2016, 37(4):44-48
-
[38]
Mitsudome T, Yamamoto M, Maeno Z, et al. J. Am. Chem. Soc., 2015, 137(42):13452-13455 doi: 10.1021/jacs.5b07521
-
[39]
Moisala A, Nasibulin A G, Kauppinen E I. J. Phys.: Condens. Matter, 2003, 15(42):3011-3035 doi: 10.1088/0953-8984/15/42/003
-
[40]
Deng B, Hsu P C, Chen G C, et al. Nano Lett., 2015, 15(6): 4206-4213 doi: 10.1021/acs.nanolett.5b01531
-
[41]
宋春蕊, 喻博闻, 邝代涛, 等.粉末冶金材料科学与工程, 2017, 22 (2):270-275SONG Chun-Rui, YU Bo-Wen, KUANG Dai-Tao, et al. Materials Science and Engineering of Powder Metallurgy, 2017, 22 (2):270-275
-
[42]
Ma L, Yu B W, Wang S L, et al. J. Nanopart. Res., 2014, 16 (8):2545-2553 doi: 10.1007/s11051-014-2545-5
-
[43]
Liu Jun, Yu B W, Zhang Q K, et al. Nanotechnology, 2015, 26(8):85601-85607 doi: 10.1088/0957-4484/26/8/085601
-
[44]
Yu B W, Wang S L, Zhang Q K, et al. Nanotechnology, 2014, 25(32):325602-325608 doi: 10.1088/0957-4484/25/32/325602
-
[45]
Liang X H, Lyon L B, Jiang Y B, et al. J. Nanopart. Res., 2012, 14:943-955 doi: 10.1007/s11051-012-0943-0
-
[46]
Liang X H, Zhou Y, Li J, et al. J. Nanopart. Res., 2011, 13: 3781-3788 doi: 10.1007/s11051-011-0299-x
-
[47]
Jiang C G, Shang Z Y, Liang X H, et al. ACS Catal., 2015, 5 (8):4814-4818 doi: 10.1021/acscatal.5b00969
-
[48]
Li P Y, Li F S, Deng G D, et al. Chem. Commun., 2016, 52 (14):2996-2999 doi: 10.1039/C5CC08848H
-
[49]
Weiher N, Beesley A, Tsapatsaris N, et al. J. Am. Chem. Soc., 2007, 129(8):2240-2241 doi: 10.1021/ja067316c
-
[50]
Zhan W C, H Q, Liu X F, et al. J. Am. Chem. Soc., 2016, 138(49):16130-16139 doi: 10.1021/jacs.6b10472
-
[51]
朱世东, 周根树, 蔡锐, 等.热处理技术与装备, 2010, 31:1-6 doi: 10.3969/j.issn.1673-4971.2010.01.001ZHU Shi-Dong, ZHOU Gen-Shu, CAI Rui, et al. Heat Treatment Technology and Equipment, 2010, 31:1-6 doi: 10.3969/j.issn.1673-4971.2010.01.001
-
[52]
Hutchings G J. Chem. Commun., 2008, 10:1148-1164
-
[53]
Anastasopoulos A, Davies J C, Hannah L, et al. ChemSusChem, 2013, 6(10):1973-1982 doi: 10.1002/cssc.v6.10
-
[54]
Reske R, Mistry H, Behafarid F, et al. J. Am. Chem. Soc., 2014, 136(19):6978-6986 doi: 10.1021/ja500328k
-
[55]
李剑, 武海顺, 杨丽娜, 等.无机化学学报, 2017, 33(4): 583-588 doi: 10.11862/CJIC.2017.072LI Jian, WU Hai-Sun, YANG Li-Na, et al. Chinese J. Inorg. Chem., 2017, 33(4): 583-588 doi: 10.11862/CJIC.2017.072
-
[56]
Bai L C, Wang X, Chen Q, et al. Angew. Chem. Int. Ed., 2016, 55(50):15656-15661 doi: 10.1002/anie.201609663
-
[57]
Li J J, Chen W, Zhao H, et al. J. Catal., 2017, 352:371-381 doi: 10.1016/j.jcat.2017.06.007
-
[58]
Reske R, Mistry H, Behafarid F, et al. J. Am. Chem. Soc., 2014, 136(19):6978-6986 doi: 10.1021/ja500328k
-
[59]
Zhang J F, Feng C, Deng Y D, et al. Chem. Mater., 2014, 26 (2):1213-1218 doi: 10.1021/cm403591g
-
[60]
Cuenya B R, Behafarid F, et al. Surf. Sci. Rep., 2015, 70: 135-187 doi: 10.1016/j.surfrep.2015.01.001
-
[61]
Ahmadi T S, Wang Z L, Green T C, et al. Science, 1996, 272:1924-1925 doi: 10.1126/science.272.5270.1924
-
[62]
Bratlie K M, Lee H, Komvopoulos K, et al. Nano Lett., 2007, 7(10):3097-3101 doi: 10.1021/nl0716000
-
[63]
Cao S W, Li Y, Zhu B C, et al. J. Catal., 2017, 349:208-217 doi: 10.1016/j.jcat.2017.02.005
-
[64]
Si R, Stephanopoulos M F. Angew. Chem. Int. Ed., 2008, 47 (15):2884-2887 doi: 10.1002/(ISSN)1521-3773
-
[65]
Singh A K, Xu Q. ChemCatChem, 2013, 5(3):652-676 doi: 10.1002/cctc.201200591
-
[66]
Wu X S, Zhao Y, Ge H B, et al. J. Am. Chem. Soc., 2015, 137(15):4924-4927 doi: 10.1021/jacs.5b01671
-
[67]
Yu C, Fu J J, Muzzio M, et al. Chem. Mater., 2017, 29(3): 1413-1418 doi: 10.1021/acs.chemmater.6b05364
-
[68]
Wang R Y, Wu Z W, Chen C M, et al. Chem. Commun., 2013, 49(74):8250-8252 doi: 10.1039/c3cc43948h
-
[69]
Wu B H, Huang H Q, Yang J, et al. Angew. Chem. Int. Ed., 2012, 51(14):3440-3443 doi: 10.1002/anie.v51.14
-
[70]
Kennedy G, Baker L R, Somorjai G A, et al. Angew. Chem. Int. Ed., 2014, 53(13):3405-3408 doi: 10.1002/anie.201400081
-
[71]
Revunova K, Nikonov G I. Dalton Trans., 2015, 44(3):840-866 doi: 10.1039/C4DT02024C
-
[72]
Fakhri P, Nasrollahzadeh M, Jaleha B, et al. RSC Adv., 2014, 4(89):48691-48697 doi: 10.1039/C4RA06562J
-
[73]
Shah D, Kaur H. J. Mol. Catal. A: Chem., 2014, 381:70-76 doi: 10.1016/j.molcata.2013.10.004
-
[74]
Fu H B, Zhang L, Wang Y, et al. J. Catal., 2016, 344:313-324 doi: 10.1016/j.jcat.2016.09.021
-
[75]
Yu L, Zhang Q, Li S S, et al. ChemSusChem, 2015, 8(18): 3029-3035 doi: 10.1002/cssc.201500869
-
[76]
Monda S, Rana U, Bhattacharjee R R, et al. RSC Adv., 2014, 4(100):57282-57289 doi: 10.1039/C4RA12080A
-
[77]
Mohanta J, Satapathy S, Si S, et al. ChemPhysChem, 2016, 17(3):364-368 doi: 10.1002/cphc.v17.3
-
[78]
顾岩, 李冠廷, 邹铖亨, 等.无机化学学报, 2017, 33(5):787-795 doi: 10.11862/CJIC.2017.102GU Yan, LIU Guan-Ting, ZOU Cheng-Heng, et al. Chinese J. Inorg. Chem., 2017, 33(5):787-795 doi: 10.11862/CJIC.2017.102
-
[79]
Yang Q H, Chen Y Z, Wang Z U, et al. Chem. Commun., 2015, 51(52):10419-10422 doi: 10.1039/C5CC03102H
-
[80]
Goksu H. New J. Chem., 2015, 39(11):8498-8504 doi: 10.1039/C5NJ01492A
-
[81]
Nasrollahzadeh M, Sajadi S M, Rostami-Vartooni A, et al. J. Colloid Interface Sci., 2016, 466:360-368 doi: 10.1016/j.jcis.2015.12.036
-
[82]
Nandi D, Siwal S, Choudhary M, et al. Appl. Catal., A, 2016, 253:31-38
-
[83]
Ye W C, Yu J, Zhou Y X, et al. Appl. Catal., B, 2016, 181: 371-378 doi: 10.1016/j.apcatb.2015.08.013
-
[84]
Ghosh B K, Hazra S, Naik B, et al. Powder Technol., 2015, 269:371-378 doi: 10.1016/j.powtec.2014.09.027
-
[85]
Feng J, Handa S, Gallou F, et al. Angew. Chem. Int. Ed., 2016, 55(31):8979-8983 doi: 10.1002/anie.201604026
-
[86]
Liu J, Cui J H, Vilela F, et al. Chem. Commun., 2015, 51 (61):12197-12200 doi: 10.1039/C5CC04476F
-
[87]
孟宪伟, 杨宏伟, 胡昌义, 等.无机化学学报, 2016, 32(11):1981-1986 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20161112&flag=1MENG Xian-Wei, YANG Hong-Wei, HU Chang-Yi, et al. Chinese J. Inorg. Chem., 2016, 32(11):1981-1986 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20161112&flag=1
-
[88]
Shah M, Guo Q X, Fu Y, et al. Catal. Commun., 2015, 65: 85-90 doi: 10.1016/j.catcom.2015.02.026
-
[89]
Dhanda R, Kidwai M, et al. J. Mater. Chem. A, 2015, 3(38): 19563-19574 doi: 10.1039/C5TA03779D
-
[90]
Nasrollahzadeh M, Jaleh B, Ehsani A, et al. New J. Chem., 2015, 39(2):1148-1153 doi: 10.1039/C4NJ01788A
-
[91]
Shukla A, Singha R K, Sasaki T, et al. Green Chem., 2015, 17(2):785-790 doi: 10.1039/C4GC01664E
-
[92]
Fang H, Wen M, Chen H X, et al. Nanoscale, 2016, 8(1):536-542 doi: 10.1039/C5NR05016B
-
[93]
Yang X L, Zhong H, Zhu Y H, et al. J. Mater. Chem. A, 2014, 2(24):9040-9047 doi: 10.1039/c4ta00119b
-
[94]
Bagal D B, Bhanage B M. Adv. Synth. Catal., 2015, 357(5): 883-900 doi: 10.1002/adsc.v357.5
-
[95]
Mukherjee A, Srimani D, Chakraborty S, et al. J. Am. Chem. Soc., 2015, 137(28):8888-8891 doi: 10.1021/jacs.5b04879
-
[96]
Lange S, Elangovan S, Cordes C, et al. Catal. Sci. Technol., 2016, 6(13):4768-4772 doi: 10.1039/C6CY00834H
-
[97]
Li Y, Gong Y T, Xu X, et al. Catal. Commun., 2012, 28:9-12 doi: 10.1016/j.catcom.2012.08.005
-
[98]
Yoshida H, Wang Yu, Narisawa S, et al. Appl. Catal., A, 2013, 456:215-222 doi: 10.1016/j.apcata.2013.03.004
-
[99]
Lu S L, Wang J Q, Cao X Q, et al. Chem. Commun., 2014, 50(26):3512-3515 doi: 10.1039/C3CC48596J
-
[100]
Marella R K, Koppadi K S, Jyothi Y, et al. New J. Chem., 2013, 37(10):3229-3235 doi: 10.1039/c3nj00453h
-
[101]
Cao Y Y, Niu LB, Wen X, et al. J. Catal., 2016, 339:9-13 doi: 10.1016/j.jcat.2016.03.015
-
[102]
Long J L, Shen K, Li Y W. ACS Catal., 2017, 7(1):275-284 doi: 10.1021/acscatal.6b02327
-
[103]
Hao C H, Guo X N, Pan Y T, et al. J. Am. Chem. Soc., 2016, 138(30):9361-9364 doi: 10.1021/jacs.6b04175
-
[104]
Wang D, Zhu YJ, Tian C G, et al. ChemCatChem., 2016, 8 (9):1718-1726 doi: 10.1002/cctc.v8.9
-
[105]
Shi J J, Zhang M Y, Du W C, et al. Catal. Sci. Technol., 2015, 5(6):3108-3112 doi: 10.1039/C5CY00393H
-
[106]
于建雁, 宋绍飞, 叶素芳, 等.无机化学学报, 2014, 30(2):271-276 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20140207&flag=1YU Jan-Yan, SONG Shao-Fei, YE Su-Fang, et al. Chinese J. Inorg. Chem., 2014, 30(2):271-276 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20140207&flag=1
-
[107]
路宁悦, 周悦, 范彬彬, 等.无机化学学报, 2015, 31(12):2324-2330 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20151207&flag=1LU Ning-Yue, ZHOU Yue, FAN Bin-Bin, et al. Chinese J. Inorg. Chem., 2015, 31(12):2324-2330 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20151207&flag=1
-
[108]
Ponec V. Appl. Catal., A, 1997, 149:27-48. doi: 10.1016/S0926-860X(96)00250-5
-
[109]
Tamura M, Yonezawa D, Oshino T, et al. ACS Catal., 2017, 7:5103-5111 doi: 10.1021/acscatal.7b01055
-
[110]
Yang L, Jiang Z S, Fan G L, et al. Catal. Sci. Technol., 2014, 4(4):1123-1131 doi: 10.1039/c3cy01017a
-
[111]
Guo Z Y, Xiao C X, Zhou L, et al. ACS Catal., 2014, 4(5): 1340-1348 doi: 10.1021/cs400982n
-
[112]
Vu K B, Konstantin V B, Dalaver H A, et al. ACS Catal., 2015, 5(4):2529-2533 doi: 10.1021/acscatal.5b00262
-
[1]
-



图 3 (A) 9.5 nm Co47Pt53 NPs的透射电镜图; (B)显示在(A)中典型的纳米粒子的高分辨透射电镜图; (C) Co47Pt53 NPs的高角度环形暗场图; (D~F) Co(红色)/Pt(绿色)的元素分布; (G) Co47Pt53 NPs高角度环形暗场图和相应的元素分布; (H) Co47Pt53 NP的EDS图[30]
Figure 3 (A) TEM image of the 9.5 nm Co47Pt53 NPs; (B) HRTEM image of a representative NP shown in (A); (C) HAADF image of four representative Co47Pt53 NPs; (D~F) Elemental mappings of Co (red)/Pt (green) signals combined (F) and single element Co (red) (D) and Pt (green) (E); (G) HAADF image and the corresponding elemental map of four representative Co47Pt53 NPs; (H) EDS line scan crossing a Co47Pt53 NP; Inset shows the NP scanned[30]






图 9 (a) 喹啉的加氢产物; (b)用不同粒径Pt作为催化剂, 喹啉的转化率和1, 2, 3, 4-四氢喹啉选择性随时间变化的曲线; (c)TOF随粒径Pt变化的曲线(每个表面积的Pt); 活化H2和D2的TOF比较, (d) Pt-1.2反应5次的转化率和选择性的比较; (d)反应前后催化剂TEM电镜图[56]
Figure 9 (a) Possible products of the quinoline hydrogenation; (b) Plots of conversion of quinoline and selectivity toward 1, 2, 3, 4-tetrahydroquinoline against reaction time with Pt-x as the catalyst; (c) Plot of TOF (per surface Pt atom) against diameter of the PtNPs. The TOF for activation of H2 and D2 to form HD is listed for comparison. Inset: Size-dependent mass activity (per unit mass of Pt) of the PtNPs in hydrogenation of quinoline; (d) Change of conversion and selectivity in 5 cycles of the catalysis with Pt-1.2 as the catalyst; (e) TEM images of the catalyst before and after cycling[56]

图 10 不同粒径Pt随时间变化甲酸产生气体的量(CO2+H2)的变化趋势
Figure 10 Plots of the volume of the generated gas (CO2+H2) versus time with five different particle sizes
Reaction condition: 5 mL solution of 0.6 mol·L-1 formic acid+0.6 mol·L-1 sodium formate in the presence of 55 mg of Pd/C (2.3%(w/w) Pd) catalysts at 25 ℃ under ambient atmosphere[57]

图 11 (a) 在1.3×104 Pa C6H6, 1.3×105 Pa H2和8.5×105Pa Ar的条件下, 十四烷基三甲基溴化铵(TTAB)稳定的Pt立方相和Pt立方八面相对环己烷和环己烯的转换率; (b)相应的阿伦尼乌斯图[62]
Figure 11 (a) Turnover rates of cyclohexane (C6H12) and cyclohexene (C6H10) formation on TTAB-stabilized cubic and cuboctahedral Pt nanoparticles (NPs) for 1.3×104 Pa C6H6, 1.3×105 Pa H2, and 8.5×105 Pa Ar, and (b) corresponding Arrhenius plots; Dotted lines are drawn as visual aides[62]


图 13 (A) Cu36Ni64 NPs的透射电镜图; (B)Cu36Ni64 NPs高分辨透射电镜图; (C)高角度环形暗场扫描透射电子显微镜图和元素分布图Ni(绿色)铜(红色); (D)氨硼烷处理的G-Cu36Ni64 NPs的透射电镜图[67]
Figure 13 (A) TEM image of the as-prepared Cu36Ni64 NPs; (B) HRTEM image of a single Cu36Ni64 NP; (C) HAADF-STEM image of the Cu36Ni64 NPs (scale bar: 10 nm) and STEM-EELS elemental mapping of a selected NP to show Ni (green) and Cu (red) distribution within the NP; (D) TEM image of the ammonia borane-treated G-Cu36Ni64 NPs[67]

图 14 (a) 不同有机胺包覆的Pt3Co催化剂在肉桂醛接近100%转化时肉桂醇(白色), 氢化肉桂醛(阴影), 氢化肉桂醇(灰色)的选择性直方图; (b)在肉桂醛完全转化时, 不同有机胺包覆的Pt3Co催化剂的氢化肉桂醇产生速率[69]
Figure 14 Histograms of (a) Selectivity for COL (white), HCAL (crosshatch), and HCOL (gray) at nearly 100% conversion in the hydrogenation of CAL with different amine-capped Pt3Co catalysts; (b) Rates of HCOL production after complete conversion of CAL with different amine-capped Pt3Co nanocatalysts[69]
Conditions: Pt3Co nanocatalysts (11 mg), n-butanol (10 mL), CAL (3 mmol), nonane (1 mmol), H2 (1.5×105 Pa), 25 ℃[69]

图 15 和频光谱图(A) Pt/SiO2和(B)Pt/TiO2在反应温度120 ℃, 1.33×102 Pa巴豆醛, 1.33×104 Pa氢气和8.92×104 Pa氩气下; 文中提到的这些光谱图是相应的表面物种[70]
Figure 15 SFG spectra for (A) Pt/SiO2 and (B) Pt/TiO2 under reaction conditions of 1.33×102 Pa crotonaldehyde, 1.33×104 Pa hydrogen, and 8.92×104 Pa argon at a catalyst temperature of 120 ℃; Assignment of these spectra, explained in the text, yields the accompanying surface intermediates[70]

图 16 (A~C)不同厚度的介孔二氧化硅包覆的金纳米棒的高分辨透射电镜: (A) Au NRs@SiO2-10, (B) Au NRs@SiO2-12, (C) Au NRs@SiO2-14; (D) Au NRs@SiO2-10的元素分布; (E)不同厚度的介孔二氧化硅包覆的金纳米棒催化对硝基苯酚的动力学常数(Au NRs@SiO2-10、AuNRs@SiO2-12和Au NRs@SiO2-14), 插图为CTAB-封装的Au NRs的动力学常数[77]
Figure 16 (A~C) HRTEM images of gold nanorods after coating with a layer of porous silica of varying shell thickness:(A) Au NRs@SiO2-10, (B) Au NRs@SiO2-12, (C) Au NRs@SiO2-14; (D) EDX elemental mapping of Au NRs@SiO2-10; (E) Kinetic data for the catalysis of 4-nitrophenol using various silica-coated gold nanorods (Au NRs@SiO2-10, AuNRs@SiO2-12, and Au NRs@SiO2-14); Inset shows the linear fitting of kinetic data with CTAB-capped Au NRs[77]








表 1 各种金属纳米复合催化剂应用于还原芳香族硝基化合物到胺基类化合物
Table 1. Summary of the reduction of aromatic nitro-compounds catalyzed by metal catalysts
 下载: 导出CSV
下载: 导出CSV
-




























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 388
- 文章访问数: 24206
- HTML全文浏览量: 5461
 下载:
下载:
