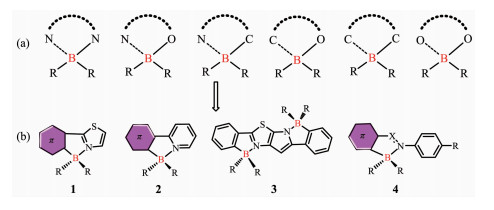
 图1
(a) 四配位硼骨架示意图; (b)N, C-螯合的四配位硼结构[22-31]
Figure1.
(a) Chelated backbones of four-coordinated organoboron compounds; (b)Structures of N, C-chelate organoboron compounds[22-31]
图1
(a) 四配位硼骨架示意图; (b)N, C-螯合的四配位硼结构[22-31]
Figure1.
(a) Chelated backbones of four-coordinated organoboron compounds; (b)Structures of N, C-chelate organoboron compounds[22-31]
引用本文:
秦妍妍, 许文娟, 胡长永, 刘淑娟, 赵强. 基于N, C螯合π共轭骨架的四配位有机硼化合物及其光电应用[J]. 无机化学学报,
2017, 33(10): 1705-1721.
doi:
10.11862/CJIC.2017.223

Citation: QIN Yan-Yan, XU Wen-Juan, HU Chang-Yong, LIU Shu-Juan, ZHAO Qiang. Four-Coordinated Organoboron Compounds with π-Conjugated N, C-Chelate Ligand and Their Optoelectronic Applications[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(10): 1705-1721. doi: 10.11862/CJIC.2017.223
Citation: QIN Yan-Yan, XU Wen-Juan, HU Chang-Yong, LIU Shu-Juan, ZHAO Qiang. Four-Coordinated Organoboron Compounds with π-Conjugated N, C-Chelate Ligand and Their Optoelectronic Applications[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(10): 1705-1721. doi: 10.11862/CJIC.2017.223
基于N, C螯合π共轭骨架的四配位有机硼化合物及其光电应用
摘要:
具有N,C-螯合π共轭骨架的四配位有机硼化合物,其分子内存在的B-N配位作用使分子骨架趋于平面,π共轭性增强,使这类化合物具有较高的电化学和热学稳定性、优异的发光性能和强的电子亲和势,成为非常有发展前景的新型光电材料,已在有机发光二极管(OLEDs)、有机场效应晶体管(OTFTs)、有机太阳能电池、传感等方面进行了广泛研究。在本文中,我们主要介绍了N,C-螯合四配位有机硼化合物的合成方法及其在电子传输材料、发光材料、光致变色材料及有机太阳能电池材料中的应用研究。
-
关键词:
- N, C-螯合四配位硼
- / 电子传输材料
- / 光致变色材料
- / 有机发光二极管
- / 有机太阳能电池
English
Four-Coordinated Organoboron Compounds with π-Conjugated N, C-Chelate Ligand and Their Optoelectronic Applications
Abstract:
Four-coordinated organoboron compounds with a π-conjugated N, C-chelate backbone have high thermal stability, intense luminescence, and strong electron affinity, and thereby, are promising candidates as novel optoelectronic materials for applications in organic light-emitting diodes (OLEDs), organic field-effect transistors (OFETs), organic solar cells and sensors. This article summarized the recent research progress on the synthesis and the application of this important class of optoelectronic materials.
-
0 引言
近年来,具有π共轭骨架的有机光电材料吸引了国内外众多学者的研究兴趣,这些材料在有机半导体器件方面具有很好的应用,已成功应用于有机太阳能电池、有机发光二极管、有机场效应晶体管、有机存储器件、化学和生物传感等方面。在碳原子骨架的大π共轭体系中引入主族元素(如硼原子),是调节有机光电信息材料性质的有效途径。通过改变硼原子与π共轭体系之间的轨道相互作用、电子效应以及杂原子价键数从而产生具有独特结构的分子,可以实现对整个分子体系电子性质的调控。近年来,具有π共轭体系的有机硼发光化合物因其独特的性能引起了化学家们的关注。这类化合物被广泛应用于非线性光学、化学传感、有机电致发光、生物分子识别等领域[1-7],我们课题组也开发了一系列含三芳基硼的金属配合物,实现了其在氟离子传感和光电器件中的应用[8-13]。
由于三配位有机硼化合物中硼原子具有空的2pπ轨道,N、O等原子的孤对电子可以填充到硼的pπ轨道中而形成配位键,从而使硼原子达到四配位。与三配位硼化合物相比,四配位硼化合物通常具有较高的化学稳定性,并且易于制备。常见的四配位有机硼化合物包括N,N-、N,O-、N,C-、C,O-、C,C-、O,O-螯合的配合物(图 1a),分子内B…X配位键的形成使分子骨架趋于平面,有利于增大π电子的离域程度,是非常有潜力的一类有机光电功能材料[14-16]。这几类四配位硼配合物中,最为知名的N,N-螯合四配位硼化合物是氟化硼配位二吡咯甲川(BODIPY)类染料,这类化合物具有高的摩尔吸光系数、良好的光稳定性、激发波长在可见光区、结构易于修饰、发射波长可调到近红外、不易受环境的影响等优点。国内外许多化学及生物学家通过在母体分子上引入不同的化学基团对其结构进行修饰,开发出许多具有不同性能BODIPY衍生物,并将这类化合物应用于各种金属离子的检测、pH值测定、DNA标记及测序等领域[17]。我们课题组[18-20]近期也制备了基于BODIPY的化合物,并实现了在离子检测中的应用。张红雨等[21]利用三苯基硼或氟化双(五氟苯基)硼烷和1,3-二芳基-β-二酮反应获得了O,O-螯合有机四配位硼化合物,其在溶液中表现出高效的绿色荧光,而在晶体中表现出红光发射,显示了聚集诱导发光颜色变化特征,且其晶体在用脉冲激光束照射时显示出明显变窄的发射,这是含硼化合物显示受激辐射放大特性的第一个例子。这一发现表明了四配位硼化合物在有机固体激光中的潜在应用,从而扩大了含硼材料的应用领域。
图1
(a) 四配位硼骨架示意图; (b)N, C-螯合的四配位硼结构[22-31]
Figure1.
(a) Chelated backbones of four-coordinated organoboron compounds; (b)Structures of N, C-chelate organoboron compounds[22-31]
基于N,C-螯合的有机四配位硼化合物主要是以结构1、2、3和4作为配体骨架构建(图 1b)。通过硼与N,C共轭骨架螯合,分子内B-N配位使分子骨架趋于平面,螯合物的π共轭性被增强,也可以有效增加材料的电子亲和势。四配位硼化合物的最低未占据分子轨道(LUMO)通常位于π共轭的螯合配体上,硼的配位使其更加趋于稳定,这使得可以用一些四配位硼化合物作为有效的电子传输材料。Yamaguchi和Wakamiya等[22-23]基于骨架1构建了一系列N,C-螯合的硼化合物,这类材料发光量子效率偏低,然而其具有低的LUMO能级和高的电子迁移率,可以作为优异的电子传输材料。四配位硼化合物的最高占有分子轨道(HOMO)可位于螯合配体或R基团上,这取决于R基团的性质。螯合物的π→π*电子跃迁或从R基团到螯合物的电荷转移跃迁, 通常决定了这类分子的发光性能。Wang等[24-28]合成了一系列基于骨架2的N,C-螯合有机硼化合物,大部分此结构的化合物具有可逆的光致变色性质,可以用于光开关。吉林大学的张红雨等[29]制备了一系列基于骨架3的双N,C-螯合的梯型有机硼化合物,这种双硼的梯型化合物具有高的热稳定性、高效的荧光发射和强的电子亲和势。Kawashima等[30-31]基于偶氮苯或亚胺类骨架4合成了一类发光的N,C-螯合有机硼化合物,共轭螯合骨架上的分子可发生π→π*跃迁。通过选择合适的螯合物和R基团,四配位硼化合物也能进行简单可逆的光热异构化,这为四配位硼的研究提供了新的机遇。
在本文中,我们总结了近年来基于N,C-螯合四配位有机硼化合物的合成方法及其光电应用。
1 合成方法
N,C-螯合四配位有机硼化合物的合成方法主要包括锂化-硼化法、金属催化法、亲电芳基硼化法及其他方法。
1.1 锂化硼化法
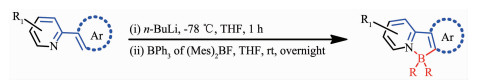
该方法是一种合成有机硼化合物的传统方法。使用卤素取代的N-杂芳香化合物,先在低温-78 ℃下和n-BuLi试剂发生锂化反应,1 h后,加入有机硼试剂低温保持1 h后再移至室温继续反应,获得相应的目标硼化产物,反应通式如图 2所示[24-29]。该反应过程复杂,所用的有机硼试剂价格昂贵,并且强亲核试剂和有机锂的碱性可能导致发生不必要的副反应,限制了结构修整的范围。
1.2 金属催化法
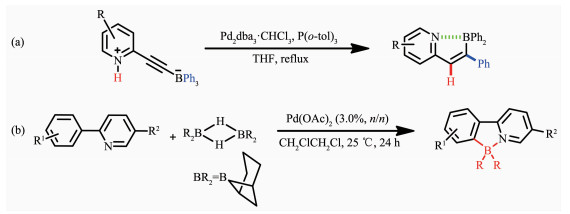
随着有机硼化合物的发展,2008年Murakami等[32]在加热的条件下, 首次使用金属钯催化含有铵盐的炔基三芳基硼烷,经过重排,得到了一种具有立体选择性的分子内含硼氮配位键的化合物。使用两性的炔基三芳基硼烷,在THF溶液中加入适量的催化剂Pd2dba3·CHCl3和P(o-tol)3,经过重排,得到了环化的含B-N配位键的化合物。该化合物表现了较强的荧光,量子效率达0.44。通式如图 3a所示。在此基础上,Kazuhiko等[33]在室温下利用金属钯催化9-borabicyclo[3.3.1]nonane(9-BBN)的重排反应合成了一种新型的N,C-螯合四配位有机硼化合物,如图 3b所示。该方法选择2-苯基吡啶衍生物作为配体骨架,利用金属钯对其进行催化,成功地以高产率获得含B-N配位键的多种氮杂环π共轭材料,并且所得的有机硼材料在紫外灯照射下可表现出有趣的发光特性[33]。有机π-共轭体系也可通过该方法进行双硼化以得到扩展。但该方法不具普遍适用性。
1.3 亲电芳香硼化法
继有机金属试剂取代反应之后,Murakami等[34]首次通过苯基吡啶及其衍生物的亲电芳基硼化反应, 合成了一系列N,C-螯合四配位有机硼化合物,且具有高的产率(图 4)。该方法比较简单且普遍适用。而且这种简单的合成方法对含硼氮配位键的氮杂π共轭材料的合成非常有用。利用这种亲电芳基硼化法,Ingleson[35]和Patil[36]分别合成了基于苯并噻二唑和喹啉的芳香硼化发光材料。Jäkle等[37]基于苯基吡啶合成了一系列梯形双硼化合物。Yam等[38]基于吡啶噻吩合成了光致变色有机硼化合物,可用于分子开关。
1.4 其它合成方法
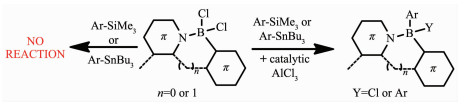
前面所述的合成方法中,锂化-硼化法得到的化合物产率较低,亲电芳香硼化法虽然可得到高产量的化合物,但这些强亲核试剂如有机铝试剂或者锌试剂却对质子酸碱敏感,产生含有离子锌盐或铝盐的混合物。因此,需要一种对空气稳定的亲核试剂,且合成的含硼化合物易分离。Ingleson等[39]在亲电芳香硼化反应的基础上发展了一种新的合成方法,即使用催化剂AlCl3,硼阳离子介导的硼氢化法对基于苯并噻二唑和芳基吡啶骨架的(N,C-螯合BCl2)进行功能化修饰。如图 5所示,在四配位硼烷(N,C-螯合BCl2)中加入AlCl3,能够生成三配位硼离子[(N,C-chelate)BCl]+。后者分别和芳基锡烷、芳基硅烷,通过硼脱甲硅基化和硼脱甲锡基化反应,生成芳基化硼烷。催化剂AlCl3的存在可以大大提高(CN-chelate)BCl2芳基化的产量。
2 N,C-螯合四配位有机硼化合物的光电应用
N,C-螯合四配位有机硼化合物由于其独特的电子结构,从而可广泛用于有机光电子领域,如作为电子传输材料、发光材料、光致变色材料以及光伏材料等用于有机发光二极管(OLEDs)、有机场效应晶体管、光敏材料和传感、有机太阳能电池等方面。
2.1 电子传输材料
在含氮π共轭化合物的适当位置引入硼原子,路易斯碱氮原子和硼原子间可形成分子内配位,这种分子内酸碱配位作用不仅以平面方式有效地固定了π共轭骨架,增加了π共轭结构的共平面性,还降低了体系的LUMO能级,使材料的带隙减小,从而有利于增强材料的电子传输特性。
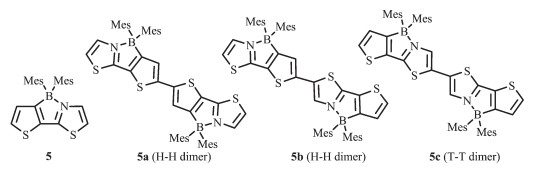
2006年,Yamaguchi等[22]基于(3-bromo-2-thienyl)-2-thiazole配体骨架,利用锂化-硼化法合成了单硼化合物5和它的双硼化合物5a~5c,这是首例四配位N,C-螯合有机硼化合物(图 6)。这类四配位有机硼化合物有较低的LUMO能级,可作为电子传输材料的一种新单元,且双硼化合物5a有相对较高的电子迁移率μ=1.5×10-4 cm2·V-1·s-1 [40], 这是由于π共轭结构的延伸和分子内的B-N配位共同作用,使得螯合物的π共轭性增强,有效地增加了材料的电子亲和势。
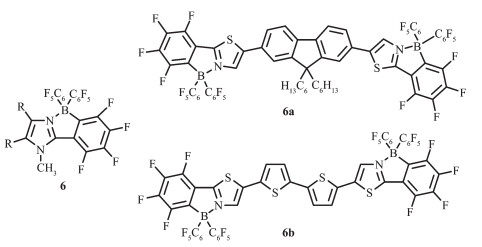
Bazan等曾报道过,路易斯酸如B(C6F5)3和含N-杂环的π共轭骨架经配位作用可进行电子调谐[41],这种修饰对控制苯并噻二唑共轭低聚物的带隙是非常有效的。噻唑π共轭骨架因有较强的吸电子能力、易进一步修饰或功能化[42]以及固态易形成紧密π堆积[43-45]等优点, 可用于n型半导体材料[42-46]。Yamaguchi等[22]先利用咪唑环和路易斯酸硼烷B(C6F5)3作用形成分子内B-N配位,再用强碱甲基锂进一步处理成环,合成化合物6,有效地降低了体系的LUMO能级,并增强吸电子性能。该团队又进一步对噻唑芴噻唑配体、噻唑联噻吩噻唑配体进行先配位再环化,得到的化合物6a和6b都有较高的热稳定性[23],Td分别在387和376 ℃(图 7)。经过配位环化后,化合物的最大吸收波长有明显的红移,这是由于共轭骨架上的分子发生π→π*跃迁,且发射光谱也发生明显的红移,6a呈现蓝光,6b呈黄光。6a和6b在正极有两步还原峰,分别为6a:Epc= -1.81 V(E1/2=-1.74 V)和-1.96 V;6b:Epc=-1.70 V(E1/2 =-1.61 V)和-1.89 V;未配位环化之前的配体还原电势分别为Epc=-2.86 V、Epc=-2.44 V,这说明配位环化过程降低了LUMO能级,这有利于电子传输器件中电子从阴极的注入。
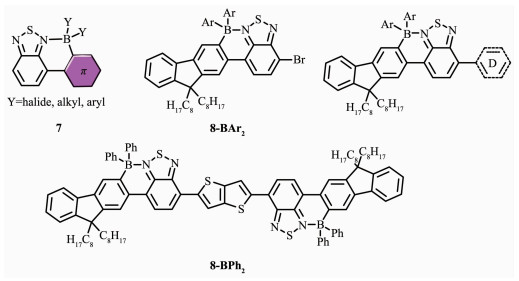
Turner等[35]使用定向C-H亲电硼氢化对含给体-受体基团的苯并噻二唑材料进行修饰,产生了一种融合的环硼材料7(图 8)。这种材料在质子溶剂(如水)中具有很好的稳定性, 吸收峰有较大红移, 还有大的Stokes位移;和未硼化之前相比,硼化后的材料7的HOMO能级基本不变,但具有显著降低的LUMO能级;且固体状态下远红外/近红外发光量子产率高达34%;溶液制造的远红外/近红外电致发光OLEDs其EQEs>0.4%。在此基础上,该团队又利用偶合反应进一步合成了硼化的9,9-二辛基芴苯并噻二唑(F8-BT)材料8-BAr2和8-BPh2[47],有效地延伸了共轭长度,使得共轭骨架更趋于平面性和刚性。这类材料也具有较低的LUMO能级、低带隙以及较高的稳定性。延伸的共轭分子8-BAr2和8-BPh2,在溶液中具有较强的吸收和发射,但在固态下其发光强度减弱,发射波长延伸到近红外区域(λmax>700 nm)且有较高的发光量子效率。如带有BPh2基团的F8-BT-F8材料,其固态荧光量子效率为33%,荧光发射在789 nm处。因此,对含D-A结构的低聚物BT化合物进行定向硼氢化,增强了材料的电子亲和势并可以调节有机半导体给受体间的带隙,这是获得具有较低LUMO能级发光材料的有效方法。
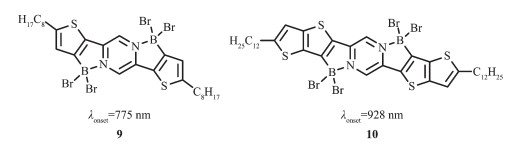
基于分子内路易斯酸碱配位作用,Fang等[48]对D-A-D结构上N进行定向硼化反应合成了低带隙共平面有机材料9和10(图 9)。该材料具有低的LUMO能级,带隙仅有1.3 eV。有趣的是,由于路易斯酸碱配位的动力学性质,这些体系的带隙可以通过外部试剂(如路易斯碱溶剂)与硼配位的竞争进行调节。这一工作为以后设计低带隙n型材料提供了模型。
2.2 发光材料
B原子引入π共轭体系后,由于其缺电子特性,可使得硼与路易斯碱如N原子形成配位作用,产生独特的电子结构和光学特性,因此可作为有应用前景的发光材料用于有机光电子领域。N,C-螯合四配位有机硼是一类新型发光材料,它们的刚性平面骨架产生了一些非常独特的性质,如强发光、高载流子迁移率和高热稳定性等。在激发光的作用下能产生强烈的荧光,可用于有机电致发光器件和生物标记等。如前文所述Ingleson和Turner等[35, 47]报道的环硼发光材料7及其延伸的π共轭分子8-BAr2和8-BPh2,其发光红移到近红外区域(λmax>700 nm)且具有较高的发光量子效率,可用于生物成像[49]。
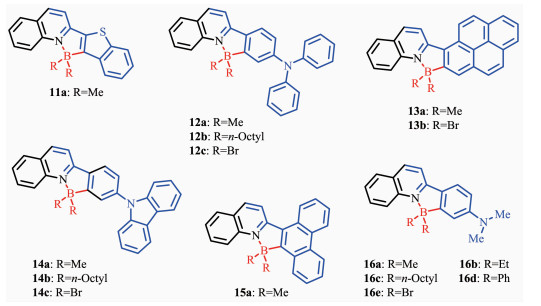
Patil等[36]报道了基于喹啉配体的芳香硼化反应制备的发光材料11a~16e(图 10)。在基于喹啉的新型N,C-螯合四配位有机硼化合物的硼位点或者N,C-螯合骨架上选取不同取代基,可以实现跨越整个可见光区域的固态发光调节。有机硼化合物11a~16e在液态和固态展现了较强的荧光。液态中所有化合物在387~491 nm有较宽的吸收带,发射带从466 nm(11a,蓝光)过渡到637 nm(12c,红光),覆盖了整个可见光区域。硼中心连有甲基的N,C-螯合四配位有机硼化合物具有较好的量子效率,如化合物13a的荧光量子效率达到0.81。所有的化合物都展现了较强的宽固态吸收(吸收峰在397~496 nm紫外可见区)和强的固态荧光发射(发射峰在491~670 nm)。如化合物11a的固态发射峰在491 nm处,呈蓝绿光;化合物16e的固态发射峰在670 nm,呈深红光。这一类材料的固态强发光性质以及可修饰性使其在生物成像和探针领域也具有潜在的应用价值。
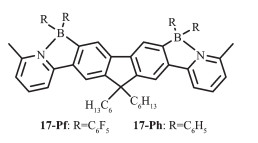
Jäkle等通过苯基吡啶二聚体的定向亲电硼化反应合成了N,C-螯合发光梯形有机硼化合物17[37](图 11)。该硼化后的梯形产物,其吸收和发射有显著的红移,并伴随着明显的电子振动结构。随着溶剂极性的增加,其吸收峰蓝移,但却不影响发射峰的位置。发光梯形有机硼化合物17呈现了较强的蓝光,具有较高的液态荧光量子效率,17-Pf可达到68%,而薄膜中的量子效率分别为7.1%(17-Ph)和3.8%(17-Pf),显著低于液态中的。化合物17-Pf的高稳定性和强蓝光可使其应用于OLEDs和成像。
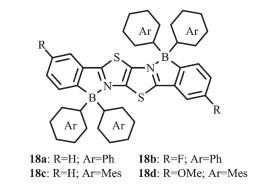
在温和的条件下,Zhang等对二苯噻唑并噻唑进行硼氢化反应合成了一系列梯形π共轭二硼化合物18a~d[29](图 12)。在骨架上引入吸电子或给电子基团,可调节分子的荧光发射。这些化合物都具有较高热稳定性并且发光在蓝绿光区域。化合物18c、d比18a、b光谱表现了明显的红移,说明在调节该类含硼梯形发光化合物的光物理特性方面,硼中心的功能化比梯形骨架的调整更有效。硼中心的大芳基取代基的空间位阻可有效保持该荧光单元分开,从而使这类材料在固态下具有强的发光性质。经过表征,合成的二硼化合物具有较高的热稳定性,较强的发光效率和强的电子亲和力,因此可作为功能材料用于有机光电器件,如OLEDs。
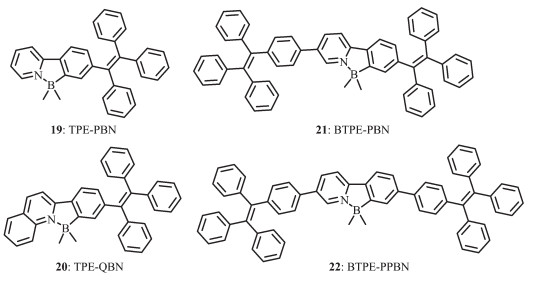
四苯乙烯(TPE)是一个有趣的螺旋桨状激活发光的特殊发光体。稀溶液中发光微弱,但在聚集形式下强烈发光,呈现聚集诱导发光(AIE)的一种新特性[50-51]。最近的研究表明,含有TPE的平面型发光团分子,聚集诱导发射,可产生高效固态发光。Qiu和Tang等[52]提出了一种比较实用的方法合成有机硼化合物,基于吡啶和喹啉设计并合成了含有TPE部分的2种新型N,C-螯合四配位有机硼化合物TPE-PBN(19)和TPE-QBN(20)(图 13),可作为有效的固态发光体。这2种新型的N,C-螯合四配位有机硼化合物具有较好的热稳定性,其分解温度分别在303和340 ℃。由于TPE的存在,2种有机硼化合物都展现了AIE特性和高聚集态发光效率。在溶液中,20的最大吸收波长在388 nm处,和没有B-N配位的配体相比,红移了57 nm;19显示了微弱的发射,没有可识别的发射峰,荧光量子效率低至0.40%。化合物20在501 nm处有较好的发射,荧光量子效率为0.94%。化合物19和20在液态呈现较弱的发射,但在聚集态却显示强蓝光发射。它们的晶体发射分别在443和462 nm,非晶形薄膜发射更为红移,分别在491和500 nm处。固态荧光量子效率分别为98%和99%,远远高于报道过的多数有机硼化合物。这说明,在聚集态时,这2种化合物有较好的AIE特性。经过理论计算和电化学研究,20的HOMO能级为-5.220 eV,和19(-5.216 eV)的比较接近,而20的LOMO(-2.108 eV)能级远低于19 (-1.719 eV)的,较低的LUMO能级有利于电子的注入与传输。
在此研究基础上,Qiu等又对基于TPE的N,C螯合四配位有机硼化合物进行了深入研究,研究了分子内B-N配位作用对其光致发光特性、电子结构、电致发光特性的影响。他们设计合成了2种新的有机硼化合物,即BTPE-PBN(21)和BTPE-PPBN(22)[53](图 13)。这2种化合物有较高的热稳定性,热分解温度分别为403和436 ℃。化合物21和22的最大吸收波长在376、373 nm,和没有配位之前相比,分别红移了27和23 nm。大部分基于TPE构建的发光材料在液态都是非荧光或者弱发光的,这是因为分子内TPE单元的自由旋转消耗了发光体激发态能量。化合物21和22在稀溶液中呈现了弱的发射,荧光量子效率分别为0.53%和0.55%,和没配位之前相比,略有升高。这是因为B-N配位减小了分子间的旋转。化合物21和22的薄膜发射峰分别位于495和490 nm,荧光量子效率达到了100%,未配位之前为81%和40%,这些数据表明,分子内B-N配位作用有利于提高发光体的发光量子效率。它们的HOMO能级和没配位之前相近,但LUMO能级分别降低为-2.87和-2.67 eV(未配位之前为-2.68和-2.54 eV),说明B-N配位作用降低了LUMO能级,有利于电子的注入和传输。和光致发光相比,化合物21和22的电致发光波长分别红移至516和520 nm。基于21的器件的最大电流效率ηC, max为4.43 cd·A-1,最大能量效率为1.64 lm·W-1,最大外量子效率ηext, max为1.52%,远远高于未配位之前的(分别为0.46 cd·A-1,0.14 lm·W-1,0.21%)。基于22的器件的最大电流效率ηC, max为4.07 cd·A-1,最大能量效率为1.68 lm·W-1,最大外量子效率ηext, max为1.43%。这些结果表明,B-N配位有利于提高电致发光器件的器件效率。这些新的固态发光N,C-螯合四配位有机硼化合物是非常有潜力的OLED发光材料。
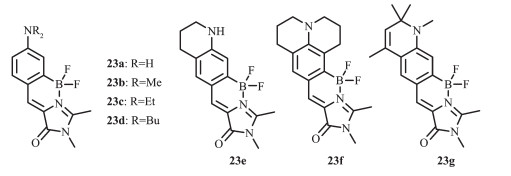
p-羟基苯亚甲基咪唑啉酮(p-HOBDI)的独特发光性能可用于RNA和蛋白的荧光探针。但由于光诱导异构化导致的低荧光量子效率,酸碱平衡引起的光谱pH值相关性,长发光波长衍生物的化学不稳定性及差的水溶性等限制了其作为荧光探针的应用。Baranov和Solntsev等[54]通过引入二氟化硼基团改善了p-HOBDI的荧光量子效率和水溶性,并成功用于荧光标记。氨基取代的p-HOBDI(即ABDI)可以使GFP发光团的光谱红移,具有显著的溶剂光致变色效应和强的氢键诱导荧光淬灭效应及抗氧化功能。因此,基于二氟化硼和氨基的结合,该团队设计合成了一种新的基于构象锁定的红移强发光GFP发光染料23[55](图 14),并研究了其不同溶剂和pH值条件下的光学性质,做了一系列同系物的光物理性质对比分析,评估了它们在荧光标记中的潜在应用。和相应的非硼化产物相比,硼化后的23a和其烷基化衍生物23b~23d的吸收和发射光谱展现了大幅度的红移,而且硼化后的产物其荧光量子效率也大幅度增加,最大达到0.82(23a)。23b~23d的荧光光谱红移的同时也伴随着显著地荧光猝灭。化合物23a具有较好的水溶性,可以更好的用于荧光探针。该团队也研究了不同氨基取代基对ABDI-BF2染料(23e~23g)荧光猝灭的影响。和23b~23d的发射光谱相比,化合物23e~23g的光谱得到了更大的红移。化合物23e表现出了和23b~23d相同的红移,由于共轭骨架的延伸,23f红移了20~30 nm,23g红移了40~50 nm,并且荧光量子效率增大到0.86(23f)。
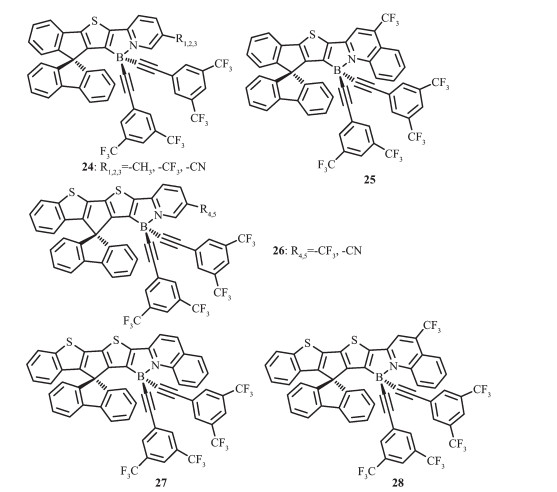
Wong等[56]设计合成了一系列对空气稳定的螺芴梯形有机硼化合物24~28(图 15),并研究了这类化合物的电化学和光物理性质。通过简单改变吡啶环上的取代基,延长螺芴骨架的π共轭结构,可以实现化合物的发光颜色从蓝到红的显著调节,覆盖了整个可见光区。该类化合物展现了宽的无结构发射峰,这是由于螺芴骨架上的噻吩基团到吡啶硼烷的分子内电荷转移跃迁导致的。除此之外,这类化合物有较高的发光量子效率,在二氯甲烷中最高可达到0.81,在薄膜中可达到0.86。其中一些化合物已被用于OLEDs中的发光材料,外量子效率EQE达到1.3%并实现了发光颜色从蓝光到红光的调节。如掺杂化合物24-R1的器件展现了蓝光发射,最大发射峰在468 nm;掺杂化合物26-R4的器件展现了绿光发射,最大发射峰在519 nm;掺杂化合物28的器件展现了红光发射,最大发射峰在605 nm。
2.3 光致变色材料
有机硼化合物也是一类新兴的光致变色材料,尤其是N,C-螯合二米基硼化合物,表现出了显著变化的光致异构化现象,且这种光致异构化过程是热可逆的,因此这类化合物可用作新型光敏材料。文献中报道过的光致变色有机硼化合物很少,只有两类,分别是Kawashima等[57]报道过的偶氮苯N,C-螯合有机硼化合物和Wang等[25]报道的N,C-螯合光致变色有机硼化合物。这两类分子都表现出了可逆的光致变色现象。其硼中心都有一个通式为BAr2(N,C-螯合)四配位四面体结构。虽然如此,这两类分子结构转变的原理却不同。一种是光诱导B-N键的解离从而引起偶氮基团的顺反异构化,另一种是基于光诱导的B-C和C-C键的断裂与形成。
2.3.1 偶氮苯N,C-螯合有机硼化合物
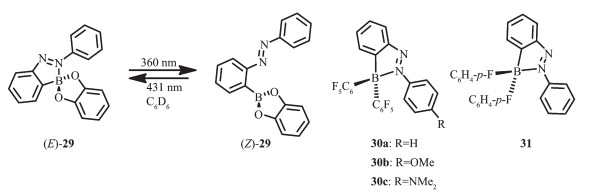
Kawashima等[57]在2005年报道了分子内含有B-N配位键的儿茶酚硼烷化合物(E)-29的光致异构化现象(图 16)。该化合物通过光照射,改变硼的配位数从而改变含偶氮基团的儿茶酚硼烷的酸性。这是因为在光照射下,化合物29会发生光致异构化过程。该分子在苯中的最大吸收波长在339 nm,这是来自于偶氮基团的π-π*跃迁。在360 nm光照射下,化合物(E)-29在339 nm处的吸收带降低,并在460 nm产生了新的吸收带,这一吸收带来自于偶氮基团的顺式异构体(Z)-29的吸收。在431 nm光照射下,顺式异构体(Z)-29可以返回到反式异构体(E)-29。这种光致异构化过程可用于调节硼的路易斯酸性,从而使得该类化合物用于分子开关。儿茶酚硼烷化合物的这种光致异构化现象和相关的二芳基硼偶氮苯化合物30和31[58-59]形成了鲜明对比。由于其较短的B-N键长(0.162 5~0.163 8 nm),在光照射下化合物30和31及它们的衍生物并不经历光致异构化过程,反而会产生较强的荧光。在正己烷溶液中,偶氮苯化合物30a和30b分别在λem=503 nm(ΦFL=0.23)和λem=524 nm(ΦFL=0.76)处显示绿光发射,而化合物30c却在λem=566 nm(ΦFL=0.52)处显示橙光发射。与之相反,化合物31并不显示发光,这可能是由于硼原子上的取代基淬灭了发光。
2.3.2 基于苯基吡啶配体的N,C-螯合有机硼化合物
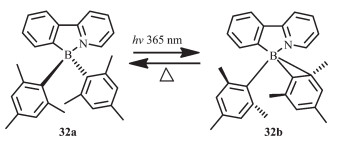
Wang等设计合成了以苯基吡啶为配体的N,C-螯合有机硼化合物32[25](图 17),在液态和固态下其结构都能保持稳定性。但是当在365 nm光照射下,该化合物的颜色可快速从无色变为深蓝色(在甲苯中,λem=458 nm,ΦFL=0.10),这是由于分子内C-C键发生了Zimmerman重排,伴随着σ键的移动;而且该过程是热可逆的,而光致异构化效率达到85%。
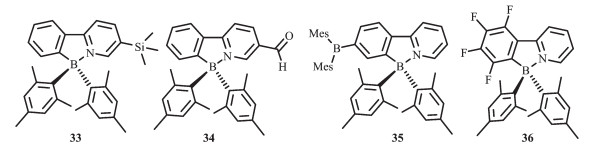
随后,他们将不同的给电子和吸电子基团引入到苯基吡啶配体上,合成了一系列N,C-螯合有机硼化合物33~36(图 18),研究不同取代基对这类硼配合物的光致变色性质的影响[25, 60]。由于πmesityl→π*ppy的电荷转移跃迁,给电子基SiMe3使得化合物发射波长蓝移(λem=418 nm,化合物33),而吸电子基-C(O)H使化合物发射波长红移(λem=~525 nm,化合物34和35),低能级吸收带向长波移动。化合物33~36都以相同的方式发生光致异构化过程,而且都伴随着显著的颜色变化。这类材料异构化后产物的吸收峰也受取代基的影响,即吸电子基使得异构化后的产物吸收峰发生红移,给电子基使之发生蓝移。这说明可通过改变取代基来实现异构化产物的颜色变化。化合物33、35的光致异构化过程都是热可逆的,而化合物34,其异构化后的产物在室温就会逐渐发生分解。化合物36不经历异构化过程,在365 nm激发时则会发生分解。
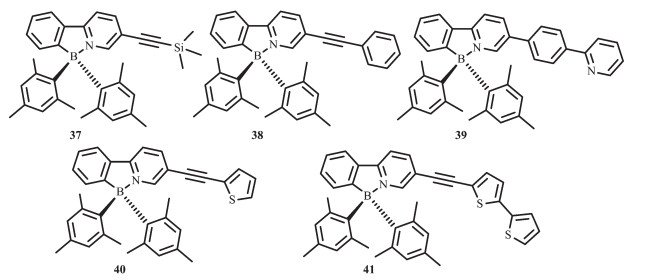
通过前面的研究发现,引入取代基来延伸螯合骨架π共轭结构可以显著影响光致异构化的量子效率和化合物的颜色。因此,Wang等又进一步合成了化合物37~41[60-61](图 19),研究了延长螯合的π共轭结构对化合物光致异构化效率和光物理性质的影响。化合物37~40都以相同的方式进行光致异构化过程,然而化合物41却不经历异构化过程。由于延伸的π共轭骨架稳定了π*能级(LUMO),这些分子的吸收和发射光谱都发生了实质性红移,其异构化产物的特征低能级吸收峰也发生了移动。这类化合物的一个重要特征就是荧光效率的显著增强。如化合物37,ΦFL=0.28,化合物38,ΦFL=0.37,化合物39,ΦFL=0.18。这表明,与光致异构化相比,荧光发射是一种更有效的激发态辐射方式。因此,这类化合物往往有更低的光致异构化量子效率,如化合物38,Φphoto=0.33,化合物39,Φphoto=0.03。π共轭结构发光效率的增强可归因于螯合骨架上π-π*跃迁的增加。化合物41证明了π共轭效应,但是化合物41并不经历如40的光致异构化过程。通过二噻吩基团引入低π→π*跃迁态,可与硼单元的电荷转移跃迁进行有效竞争,可能引起了这种不寻常的现象。因此将二噻吩基团掺入含BMes2 π共轭化合物中,可有效提高材料的光稳定性,防止它通过异构化降解。
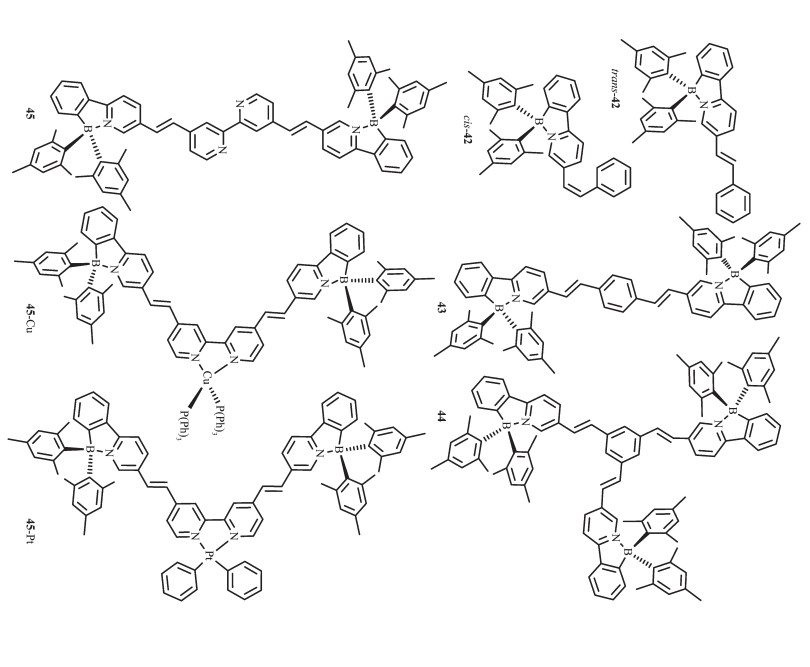
为检测烯键对N,C-螯合有机硼化合物的光致开关的影响,Wang等设计合成了化合物42~44[26],如图 20所示。这些分子发蓝或蓝绿光,溶液中ΦFL=0.30~0.59,固态约为0.23。365 nm激发时,没有颜色变化。但反式化合物42的吸收光谱有明显的蓝移,主要吸收带在370 nm处,这是烯键顺反异构的标志。对多硼化合物43和44来说,只有一个烯键发生顺反异构。然而,一个单烯键的异构化已足够关闭硼中心的光致异构化过程,稳定B(ppy)Mes2发光团。该团队合成的化合物45,在365 nm处激发时,烯键会发生光致异构化过程,当在2,2′-联吡啶位置引入Cu(PPh3)2+后[62],金属离子对烯键的异构化过程没有显著影响,但是引入PtPh2后,金属离子完全抑制了化合物45烯键的光致异构化过程,这是由于低MLCT态的存在,有效地拦截了激发态能级,因此展示了烯键异构化。这说明分子内烯键和低的MLCT态都有利于硼发光团的稳定性。
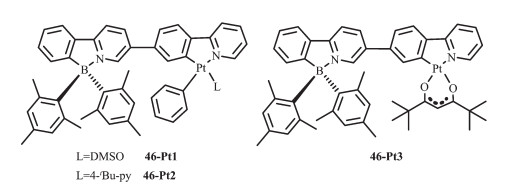
Wang等[61]在基于苯基吡啶配体的N,C-螯合有机硼化合物中又进一步引入重金属铂配位获得配合物46(图 21),研究了金属离子的螯合或者简单配位对B(ppy)Mes2光致变色的影响。室温下这些化合物在甲苯中发磷光,而且都有较高的量子效率(0.13~0.45)。此Pt配合物的磷光源于螯合配体的3LC态。金属Pt的螯合增强了配体中心单线态到三线态的系间蹿跃,增强了分子的磷光。而且发现这3种Pt配合物中的B(ppy)Mes2发光团都可发生光致异构化过程,但异构化效率较低,这是由于Pt配合物的高效磷光抑制了B(ppy)Mes2发光团的光致异构化过程。
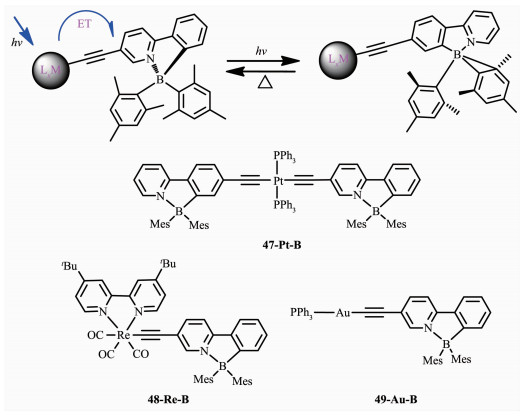
为进一步理解三线态的作用,Wang等又研究了Re(Ⅰ)([Re(CO)3(tert-Bu2bpy)]+)和Au(Ⅰ)([Au(PPh3)]+)和Pt(Ⅱ)([Pt(PPh3)2]+2)对B(ppy)Mes2异构化的影响[63]。选择这三种金属单元是因为它们独特的三线态能级和配位结构(图 22)。这些化合物的吸收光谱在270~430 nm之间,甲苯溶液(1×10-5 mol·L-1)中的发射光谱在400~700 nm。在室温390 nm激发,化合物47-Pt-B呈现双重发射,分别在480和520 nm处。时间分辨磷光光谱和氧敏感试验证明520 nm处的峰是磷光,480 nm的峰是荧光,而且2种峰都是源于共轭的B(ppy)Mes2单元。77 K时,47-Pt-B的最大磷光发射峰在516 nm。室温388 nm激发,化合物48-Re-B在615 nm有一宽的无特征发射峰,77 K时48-Re-B在520 nm表现了很强的发光,是来自骨架中B(ppy)Mes2的磷光。从室温到77 K,磷光发射带的巨大蓝移,是由骨架上3π-π*高效发射导致的。室温365 nm激发,化合物49-Au-B在490 nm处有一宽峰,在500 nm处有一尖峰。时间分辨磷光光谱表明490 nm处的是荧光,500 nm处的是磷光。365 nm照射2 h,48-Re-B没有任何光致异构化现象,这是由于Re(Ⅰ)较低的MLCT/CT态有效地猝灭硼发光团的异构化过程,提高了化合物48-Re-B的稳定性。相比之下,49-Au-B经历了一个快速的热可逆异构化过程,定量形成了其异构体,颜色从无色变为深蓝色,吸收光谱在620 nm处有一新的宽吸收带。室温365 nm激发,49-Au-B异构化效率为0.46,显著低于B(ppy)Mes2的(ΦPI=0.85)。在365 nm激发时化合物47-Pt-B也发生光致异构化过程,但是和49-Au-B相比,其光致异构化速率很慢且量子效率低至0.001,溶液颜色和紫外可见吸收光谱的变化都比较小,在600 nm处有弱的宽峰。这是由于2个B(ppy)Mes2单元共轭螯合骨架较低的π→π*态导致的。TD-DFT计算和3种含金属硼化合物的光谱研究证实,B(ppy)Mes2的光致异构化可通过三重光活性态进行,并且具有合适三线态能级的金属单元可以被用来调整该体系。
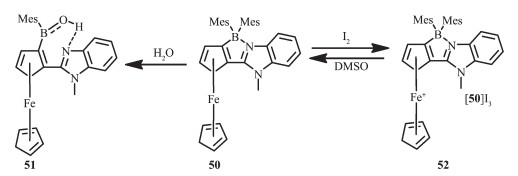
二茂铁是调节光电开关体系最常用的金属单元。为了检验二茂铁单元和N,C-螯合BMes2单元在电子、化学和光化学性质的相互影响,Wang等设计并合成含二茂铁单元的N,C-螯合四配位硼化合物50[64](图 23)。化合物50显示了一个不同寻常的动态交换和开环水解特性。水解产物(51)比较稳定,这是因为在溶液中,B-N键断裂后形成三配位硼,氮原子与OH形成氢键,稳定了化合物。50的易水解行为是不含二茂铁的N,C-螯合四配位米基硼所没有的。化合物50和51具有独特的电子特性,低能级二茂铁的特征吸收带分别在490 nm (1)和450 nm (2),摩尔吸收系数为1 200 L·mol-1·cm-1,所以这2种化合物显示了独特的颜色。化合物50中加入温和的氧化剂如I2就可得到深棕色的物种[50]I3(52)。观察其紫外可见近红外吸收光谱,有一宽吸收带在600~1 300 nm范围内,且最大吸收波长在950 nm,摩尔吸收系数为400 L·mol-1·cm-1。这个低能级带是配体到金属的电荷转移跃迁产生的。氮气环境中,光激发或者白光照射数小时,化合物50都很稳定,不会发生任何变化。硼单元对光致异构化现象的不活泼性是由于二茂铁单元的低能级电子态通过束缚激子,有效地猝灭了其光致异构化过程,而且二茂铁的大体积空间位阻和螯合骨架导致了B-N键的分解,实现了硼开环水解的动态交换。固态化合物52在室温稳定,但在弱极性溶剂如DMSO中可以还原回到化合物50。由于52中低的LMCT/d-d态,延缓了硼中心的光致异构化过程。研究发现,液态化合物52对光敏感,会快速分解,但并未观察到硼中心的光致异构化现象。
2.3.3 基于芳香杂环配体的N,C-螯合有机硼化合物
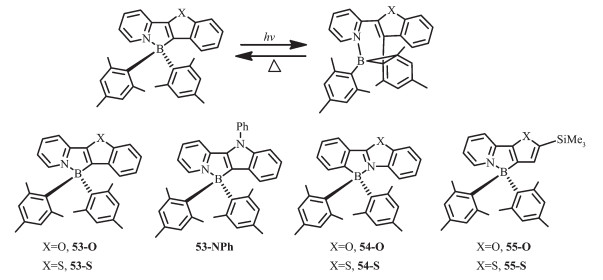
为了验证观察到的基于ppy配体N,C-螯合的有机硼化合物的光致异构现象是否是一普遍现象,Wang等设计合成了一些基于芳香杂环的有机硼化合物53~55[60, 65-67],杂环基团如苯并呋喃、苯并噻吩、N-苯基吲哚代替了ppy中的苯环(图 24)。化合物53-O、53-S、55-O、55-S都发蓝光(λem=450 nm),ΦFL=0.30 ~0.80。化合物53-NPh发蓝绿光(λem=490 nm),ΦFL=0.32。在365 nm激发,化合物53都以32a相同的方式异构化为深色异构体,如53-O是深蓝色;53-S是深绿色;53-NPh是深蓝绿色。化合物53异构化后的产物其吸收光谱和32a相比,发生了弱微红移,光致异构化量子效率低至0.10。相比之下,化合物55-O和55-S有较高的光致异构化量子效率,分别为0.35和0.75。化合物54也可发生光致异构化过程,产生深色异构体,但这类异构体对热不稳定,会进一步发生结构变化。
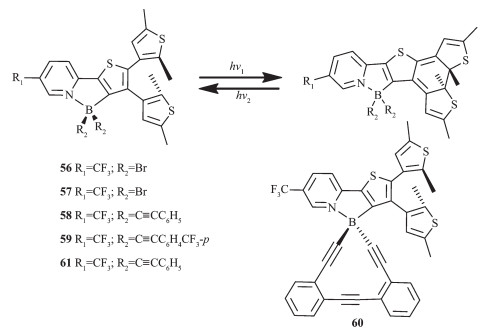
在此前的研究基础上,Yam等设计合成了一系列基于噻吩吡啶双(炔基)骨架的有机硼光致变色配合物56~61[38],结构如图 25所示。该类化合物由于吸电子基炔基的存在,降低了化合物的吸收能级,光激发时,发射带在460~580 nm,荧光量子效率在0.05~0.38之间。光致异构化也导致了发光强度的降低,这是由于闭环较低的激发态以能量转移的方式猝灭了荧光。
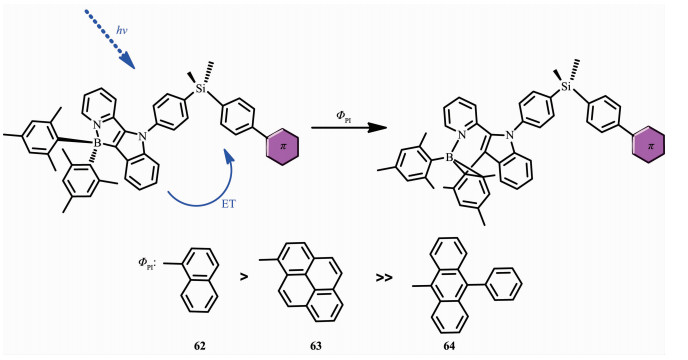
之前的研究表明,大量的光致变色材料包括螺吡喃、螺嗪和二芳基乙烯,是通过光敏三线态发生异构化的。为了证明有机硼烷中光敏激发态的本质,Wang和Yamaguchi等将光致变色硼单元和不同三线态能级的受体发光团结合,设计合成了一系列化合物62~64[68](图 26)。受体发光团苯基萘(62)、苯芘(63)、联苯蒽(64)单线态能级分别为3.93、3.33、3.05 eV,都高于硼单元的(2.77 eV)。所有的化合物的最大吸收峰在417 nm,并有相似的消光系数,归因于混合的π-π*跃迁和硼单元电荷转移跃迁,使得化合物在固态和液态都呈浅黄色,发射光谱在λem=498~501 nm,呈黄绿光。但是,由于受体发光团的存在导致了化合物62~64有不同的光致变色行为。在419 nm处,甲苯溶液(10-5 mol·L-1)中激发,化合物62和63都发生了定量的转变,光致异构化量子效率分别为ΦPI=0.12,0.13。化合物64的异构化过程完全被禁止了,没有观察到任何光谱变化。此外,样品的光反应活性可以通过有效的调节光致变色单元和三重态受体发色团之间的三线态能隙。这一发现在光致变色N,C-螯合硼化合物的设计具有重要意义,可以使用适当的三重态敏化剂或受体来敏化或猝灭BMes2单元的光异构过程。
2.4 太阳能电池材料
N,C-螯合四配位π共轭有机硼化合物具有较低的LUMO/HOMO能隙和较高的电子迁移率,因此这类材料也可以通过修饰作为有机太阳能电池的受体材料。
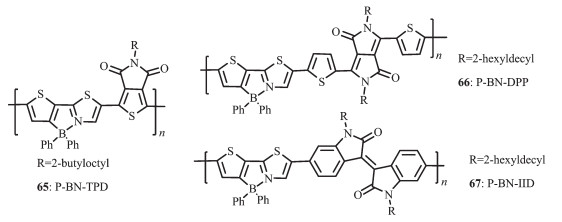
Yamaguchi等[22]发展了一种含B-N单元的新的噻吩噻唑骨架以设计合成具有高电子迁移率的电子传输小分子。Wang等[69-70]发展了一种新的方法来制备聚合物电子受体材料65(图 27),即将传统的聚合物电子给体材料中的C-C单元利用B-N单元进行取代,这种方法可以明显降低共轭聚合物的LUMO和HOMO能级,约0.6 eV。这次工作不仅提供了一种有效的方法调节共轭聚合物的LUMO和HOMO能级,而且利用有机硼化学作为一种新的方法发展具有高电子亲和力的共轭聚合物,应用于聚合物光电器件。尽管得到的聚合物P-BN-TPD(65)展现了低ELUMO/HOMO,但是由于其较低的电子迁移率,其all-PSC器件性能却很差。为了进一步提高这一类聚合物的电子迁移率,他们合成了聚合物P-BN-DPP(66)和P-BN-DPP(67)(图 27),与原来的聚合物P-BN-TPD(65)相比,增加了聚合物电子受体重复单元的长度,这样可以减轻大体积苯基的位阻效应,从而有利于聚合物骨架的π-π堆积,使材料的电子迁移率增强了近2个数量级,P-BN-IID、P-BN-DPP、P-BN-TPD的电子迁移率分别为2.80×10-5、8.48×10-6、3.40×10-7 cm2·V-1·s-1。聚合物作电子受体的all-PSCs的能量转换效率从0.12%增加到5.04%。
3 结论和展望
本文中,我们总结了N,C-螯合四配位有机硼化合物的合成方法及其在电子传输材料、发光材料、光致变色材料及有机太阳能电池材料中的应用研究。这类有机硼化合物具有丰富的化学、物理特性及多功能应用,是一类非常具有发展潜力的有机光电功能材料。这类化合物中的分子内B-N配位作用在稳定螯合配体、增强π共轭性和影响π体系的电子特性等方面起到了重要的作用,在发光材料和电子传输材料领域具有很好的潜在应用价值。此外,光致变色N,C-螯合四配位硼化合物为研究发展光致变色材料及其应用提供了新的机遇。许多新的有机硼化合物具有颜色易调节、强发光以及高光致异构化量子效率等特点,这使得它们可应用于光致变色器件。但是在聚合物体系中,这种光致变色化合物的光致异构化过程及稳定性研究的还比较少,仍需深入的评估其在光致变色器件中的可行性,而且含N,C-螯合四配位硼单元的共轭聚合物作为聚合物太阳能电池的电子受体材料的研究还很少,值得进一步的深入研究。目前,有机硼材料的研究主要集中于三配位硼和四配位硼化合物,这两类化合物都具有强发光和高电子亲和性,因此,将这两种基团引入同一个分子中,将会产生更有趣的特性,这类分子目前研究得还很少。
-
-
[1]
Sun Y, Ross N, Zhao S B, et al. J. Am. Chem. Soc., 2007, 129:7510-7511 doi: 10.1021/ja0725652
-
[2]
Gale P A. Acc. Chem. Res., 2006, 39:465-475 doi: 10.1021/ar040237q
-
[3]
Mazzeo M, Vitale V, DellaSala F, et al. Adv. Mater., 2005, 17:34-39 doi: 10.1002/(ISSN)1521-4095
-
[4]
Lin S L, Chan L H, Lee R H, et al. Adv. Mater., 2008, 20:3947-3952 doi: 10.1002/adma.v20:20
-
[5]
Noda T, Shirota Y. J. Am. Chem. Soc., 1998, 120:9714-9715 doi: 10.1021/ja9817343
-
[6]
Liu Z Q, Fang Q, Wang D, et al. Chem. Eur. J., 2003, 9:5074-5084 doi: 10.1002/(ISSN)1521-3765
-
[7]
Hudson Z M, Wang S. Dalton Trans., 2011, 40:7805-7816 doi: 10.1039/c1dt10292c
-
[8]
Zhao Q, Li F Y, Liu S J, et al. Inorg. Chem., 2008, 47:9256-9264 doi: 10.1021/ic800500c
-
[9]
Xu W J, Liu S J, Zhao X Y, et al. Chem. Eur. J., 2010, 16:7125-7133 doi: 10.1002/chem.201000362
-
[10]
Xu W J, Liu S J, Ma T C, et al. J. Mater. Chem., 2011, 21:13999-14007 doi: 10.1039/c1jm11987g
-
[11]
Xu W J, Liu S J, Sun H B, et al. J. Mater. Chem., 2011, 21:7572-7581 doi: 10.1039/c1jm00071c
-
[12]
Xu W J, Liu S J, Zhao X, et al. Chem. Eur. J., 2013, 19:621-629 doi: 10.1002/chem.201202827
-
[13]
Lin W P, Tan Q, Liang H, et al. J. Mater. Chem. C, 2015, 3:1883-1887 doi: 10.1039/C4TC02457E
-
[14]
Li D, Zhang H Y, Wang Y. Chem. Soc. Rev., 2013, 42:8416-8433 doi: 10.1039/c3cs60170f
-
[15]
Tanaka K, Chujo Y. NPG Asia Mater., 2015, 7:e223-e238 doi: 10.1038/am.2015.118
-
[16]
Rao Y L, Wang S. Inorg. Chem., 2011, 50:12263-122275 doi: 10.1021/ic200658v
-
[17]
Lu H, Mack J, Yang Y C, et al. Chem. Soc. Rev., 2014, 43:4778-4823 doi: 10.1039/C4CS00030G
-
[18]
Sun H B, Dong X C, Liu S J, et al. J. Phys. Chem. C, 2011, 115:19947-19954 doi: 10.1021/jp206396v
-
[19]
Liu S J, Shi Z J, Xu W J, et al. Dyes Pigm., 2014, 103:145-153 doi: 10.1016/j.dyepig.2013.12.004
-
[20]
Sun H B, Dong X C, Liu S J, et al. J. Phys. Chem. C, 2011, 115:19947-19954 doi: 10.1021/jp206396v
-
[21]
Wang L, Zhang Z Y, Cheng X, et al. J. Mater. Chem. C, 2015, 3:499-505
-
[22]
Wakamiya A, Taniguchi T, Yamaguchi S. Angew. Chem. Int. Ed., 2006, 45:3170-3173 doi: 10.1002/(ISSN)1521-3773
-
[23]
Job A, Wakamiya A, Kehr G, et al. Org. Lett., 2010, 12:5470-5473 doi: 10.1021/ol102282x
-
[24]
Baik C, Murphy S K, Wang S, et al. Org. Lett., 2010, 12:5266-5269 doi: 10.1021/ol102319t
-
[25]
Rao Y L, Amarne H, Zhao S B, et al. J. Am. Chem. Soc., 2008, 130:12898-12900 doi: 10.1021/ja8052046
-
[26]
Baik C, Hudson Z M, Amarne H, et al. J. Am. Chem. Soc., 2009, 131:14549-14559 doi: 10.1021/ja906430s
-
[27]
Baik C, Murphy S K, Wang S. Angew. Chem. Int. Ed., 2010, 49:8224-8227 doi: 10.1002/anie.v49:44
-
[28]
Amarne H, Baik C, Murphy S K, et al. Chem. Eur. J., 2010, 16:4750-4761 doi: 10.1002/chem.200903582
-
[29]
Li D, Zhang Z L, Zhang S S, et al. Dalton Trans., 2011, 40:1279-1285 doi: 10.1039/c0dt01269f
-
[30]
Yoshino J, Furuta A, Kambe T, et al. Chem. Eur. J., 2010, 16:5026-5035 doi: 10.1002/chem.v16:17
-
[31]
Yoshino J, Kano N, Kawashima T. J. Org. Chem., 2009, 74:7496-7503 doi: 10.1021/jo901733b
-
[32]
Ishida N, Narumi M, Murakami M. Org. Lett., 2008, 10:1279-1281 doi: 10.1021/ol8001305
-
[33]
Kuninobu Y, Iwanaga T, Omura T, et al. Angew. Chem. Int. Ed., 2013, 52:4431-4434 doi: 10.1002/anie.201210328
-
[34]
Ishida N, Moriya T, Goya T, et al. J. Org. Chem., 2010, 75:8709-8712 doi: 10.1021/jo101920p
-
[35]
Crossley D L, Cade I A, Clark E R, et al. Chem. Sci., 2015, 6:5144-5151 doi: 10.1039/C5SC01800E
-
[36]
Shaikh A C, Ranade D S, Thorat S, et al. Chem. Commun., 2015, 51:16115-16118 doi: 10.1039/C5CC06351E
-
[37]
Yusuf M, Liu K L, Guo F, et al. Dalton Trans., 2016, 45:4580-4587 doi: 10.1039/C5DT05077D
-
[38]
Wong H L, Wong W T, Yam V W W. Org. Lett., 2012, 14:1862-1865 doi: 10.1021/ol3004595
-
[39]
Crossley D L, Cid J, Curless L D, et al. Organometallics, 2015, 34:5767-5774 doi: 10.1021/acs.organomet.5b00857
-
[40]
Yasuda T, Yamaguchi Y, Zou D C, et al. J. Appl. Phys., 2002, 41:5626-5629 doi: 10.1143/JJAP.41.5626
-
[41]
Welch G C, Coffin R, Peet J, et al. J. Am. Chem. Soc., 2009, 131:10802-10803 doi: 10.1021/ja902789w
-
[42]
Jiang P, Morales G M, You W, et al. Angew. Chem. Int. Ed., 2004, 43:4471-4475 doi: 10.1002/(ISSN)1521-3773
-
[43]
Curtis M D, Cao J, Kampf J W. J. Am. Chem. Soc., 2004, 126:4318-4328 doi: 10.1021/ja0397916
-
[44]
Yamamoto T, Arai M, Kokubo H. Macromolecules, 2003, 36:7986-7993 doi: 10.1021/ma030167n
-
[45]
Mamada M, Nishida J I, Kumaki D, et al. Chem. Mater., 2007, 19:5404-5409 doi: 10.1021/cm071505s
-
[46]
Hong X M, Katz H E, Lovinger A J, et al. Chem. Mater., 2001, 13:4686-4691 doi: 10.1021/cm010496z
-
[47]
Crossley D L, Vitorica-Yrezabal I, Humphries M J, et al. Chem. Eur. J., 2016, 22:12439-12448 doi: 10.1002/chem.v22.35
-
[48]
Zhu C Z, Guo Z H, Mu A U, et al. J. Org. Chem., 2016, 81:4347-4352 doi: 10.1021/acs.joc.6b00238
-
[49]
Pansare V J, Hejazi S, Faenza W J, et al. Chem. Mater., 2012, 24:812-827 doi: 10.1021/cm2028367
-
[50]
Hong Y, Lam J W Y, Tang B Z, et al. Chem. Soc. Rev., 2011, 40:5361-5388 doi: 10.1039/c1cs15113d
-
[51]
Zhao Z J, Wang Z M, Lu P, et al. Angew. Chem. Int. Ed., 2009, 48:7608 doi: 10.1002/anie.v48:41
-
[52]
Zhao Z J, Chang Z F, He B R, et al. Chem. Eur. J., 2013, 19:11512-11517 doi: 10.1002/chem.v19.35
-
[53]
He B R, Chang Z F, Jiang Y B, et al. Dyes Pigm., 2014, 101:247-253 doi: 10.1016/j.dyepig.2013.10.014
-
[54]
Baranov M S, Lukyanov K A, Borissova A O, et al. J. Am. Chem. Soc., 2012, 134:6025-6032 doi: 10.1021/ja3010144
-
[55]
Baranov M S, Solntsev K M, Baleeva N S, et al. Chem. Eur. J., 2014, 20:13234-13241 doi: 10.1002/chem.201403678
-
[56]
Wong B Y W, Wong H L, Wong Y C, et al. Chem. Eur. J., 2016, 22:1-13 doi: 10.1002/chem.201504553
-
[57]
Kano N, Yoshino J, Kawashima T. Org. Lett., 2005, 7:3909-3911 doi: 10.1021/ol051337e
-
[58]
Yoshino J, Kano N, Kawashima T. Chem. Lett., 2008, 37:960-961 doi: 10.1246/cl.2008.960
-
[59]
Yoshino J, Kano N, Kawashima T. Chem. Commun., 2007, 6:559-561
-
[60]
Amarne H, Baik C, Wang S. Chem. Eur. J., 2010, 16:4750-4761 doi: 10.1002/chem.200903582
-
[61]
Rao Y L, Wang S. Organometallics, 2011, 30:4453-4458 doi: 10.1021/om200534m
-
[62]
Baik C, Wang S. Chem. Commun., 2011, 47:9432-9434 doi: 10.1039/c1cc13054d
-
[63]
Wang N, Ko S B, Lu J S, et al. Chem. Eur. J., 2013, 19:5314-5323 doi: 10.1002/chem.201204048
-
[64]
Rao Y L, Kusamoto T, Sakamoto R, et al. Organometallics, 2014, 33:1787-1793 doi: 10.1021/om500138f
-
[65]
Rao Y L, Amarne H, Chen L D, et al. J. Am. Chem. Soc., 2013, 135:3407-3410 doi: 10.1021/ja400917r
-
[66]
Amarne H, Baik C, Wang R Y, et al. Organometallics, 2011, 30:665-668 doi: 10.1021/om101111p
-
[67]
Rao Y L, Amarne H, Lu J S, et al. Dalton Trans., 2013, 42:638-644 doi: 10.1039/C2DT31370G
-
[68]
Hudson Z M, Ko S B, Yamaguchi S, et al. Org. Lett., 2012, 14:5610-5613 doi: 10.1021/ol302742g
-
[69]
Dou C D, Ding Z C, Zhang Z J, et al. Angew. Chem. Int. Ed., 2015, 54:3648-3652 doi: 10.1002/anie.201411973
-
[70]
Zhao R Y, Dou C D, Xie Z Y, et al. Angew. Chem. Int. Ed., 2016, 55:5313-5317 doi: 10.1002/anie.201601305
-
[1]
-












-

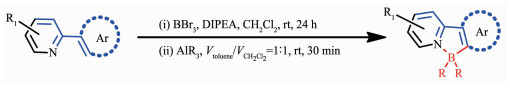

 下载:
下载:


























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 84
- 文章访问数: 13485
- HTML全文浏览量: 1605
 下载:
下载:
