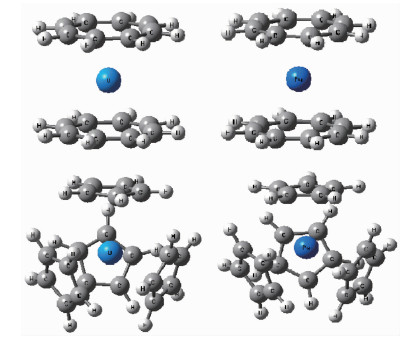
 图1
环辛四烯(COT2-)和环戊二烯(Cp-)的金属铀和钚有机化合物的结构(Cp4An和COT2An)
Figure1.
Structures of cyclopentadienyl (Cp-) and annulene(COT2-) sandwiched uranium and plutonium compounds (Cp4An and COT2An)
图1
环辛四烯(COT2-)和环戊二烯(Cp-)的金属铀和钚有机化合物的结构(Cp4An和COT2An)
Figure1.
Structures of cyclopentadienyl (Cp-) and annulene(COT2-) sandwiched uranium and plutonium compounds (Cp4An and COT2An)
引用本文:
辜家芳, 许可, 陈文凯. Cp4An和COT2An(Cp-=C5H5-, COT2-=C8H82-, An=U(Ⅳ), Pu(Ⅳ))的结构和光谱性质的密度泛函理论研究[J]. 无机化学学报,
2017, 33(9): 1579-1586.
doi:
10.11862/CJIC.2017.179

Citation: GU Jia-Fang, XU Ke, CHEN Wen-Kai. DFT Studies on Structure and Spectral Properties of Organoactinide Complexes of Cp4An and COT2An (Cp-=C5H5-, COT2-=C8H82-, An=U(Ⅳ), Pu(Ⅳ))[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(9): 1579-1586. doi: 10.11862/CJIC.2017.179
Citation: GU Jia-Fang, XU Ke, CHEN Wen-Kai. DFT Studies on Structure and Spectral Properties of Organoactinide Complexes of Cp4An and COT2An (Cp-=C5H5-, COT2-=C8H82-, An=U(Ⅳ), Pu(Ⅳ))[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(9): 1579-1586. doi: 10.11862/CJIC.2017.179
Cp4An和COT2An(Cp-=C5H5-, COT2-=C8H82-, An=U(Ⅳ), Pu(Ⅳ))的结构和光谱性质的密度泛函理论研究
摘要:
采用密度泛函理论研究气相和四氢呋喃(THF)溶剂中Cp4An和COT2An(Cp-=C5H5-,COT2-=C8H82-,An=U(Ⅳ),Pu(Ⅳ))配合物的性质。THF溶剂对配合物的溶剂化效应采用类导体极化连续模型(CPCM)近似计算。计算结果显示在THF溶液中各配合物结合能的大小顺序为COT2Pu > COT2U > Cp4Pu > Cp4U。溶剂化效应降低了该金属有机配合物的结合能。计算得到的化合物的结构参数和红外光谱数据与实验数据保持一致。通过对Cp4An和COT2An(An=U(Ⅳ),Pu(Ⅳ))的分子轨道能级图分析发现,采用最高的RSC ECP赝势计算COT2U和Cp4U的基态分别为三重fφ2和fσ2组态;而COT2Pu和Cp4Pu的基态分别为五重fσ1fπ1fφ1和fσ3fδ1组态。
English
DFT Studies on Structure and Spectral Properties of Organoactinide Complexes of Cp4An and COT2An (Cp-=C5H5-, COT2-=C8H82-, An=U(Ⅳ), Pu(Ⅳ))
Abstract:
Properties of Cp4An and COT2An (Cp-=C5H5-, COT2-=C8H82-, An=U(Ⅳ), Pu(Ⅳ)) species both in gas phase and tetrahydrofuran (THF) solution are systematically studied by DFT (density functional theory). The solvent effect of THF is simulated by a conductor-like polarized continuum model (CPCM). Theoretical calculations show that the binding energies of the complexes are in the order of COT2Pu>COT2U>Cp4Pu>Cp4U. Solvent effects decreased binding energies of the structures. The optimized geometry structures and the IR spectra for the complexes in gas and aqueous phase are in accordance to the available experimental data very well. Detail studies on MO energy levels for Cp4An and COT2An with An=U(Ⅳ), Pu(Ⅳ)calculated with rsc ecp indicate that COT2U and CP4U favors triplets fφ2 and fσ2 configuration respectively, meanwhile, COT2Pu and Cp4Pu favor quintets fσ1fπ1fφ1 and fσ3fδ1 configuration respectively.
-
Key words:
- organometallic
- / uranium
- / plutonium
- / density functional theory
- / solvent effect
-
0 引言
锕系元素U和Pu在元素周期表中的地位特殊,U和Pu的外层价电子组态分别是5f36d17s2和5f67s2,锕系元素特殊的电子组态,表现在它们和配体配位时情况变得比一般的过渡金属更加复杂;其次,由于它们是重原子,核外电子存在相对论效应,量子化学方法处理起来也较为困难。在地下水中,由于锕系离子可以与水中的多种无机配体形成配合物的形式进行迁移而倍受实验和理论工作者的关注[1-6]。本课题组先前应用密度泛函理论研究了铀与多种无机配体配位的性质,并取得一定的成果[7-9]。锕系元素也能与有机配体形成特殊的结构,例如锕系离子铀环辛四烯(COT2-)和环戊二烯(Cp-)的金属铀有机配合物具有类似二茂铁的夹心结构。1986年合成出的二环辛四烯铀(COT2U)引起理论和实验科学家的重视。理论上主要是针对气相下该类有机配合物的结构进行研究,而关于液相下的结构研究很少[10-24]。COT2Pu和COT2U是在四氢呋喃溶液中,通过将固体[(C2H5)4N2]PuCl5和Py2UCl6加入到K2COT的四氢呋喃溶液中合成出来[25-27]。Cp4U则是通过UC14与KC5H5在苯溶液中反应制备得到的[28-31]。Fischer等[28]发现合成得到的红色晶体状的Cp4U在空气中能够稳定存在,但在苯溶液中的结构受空气影响而不稳定。实验上也得到了Cp4U的X射线晶体结构数据[29-30]。因此考虑溶剂化效应对结构稳定性的影响具有重要的意义。
理论上应用相对论lcao Hartree-Fock-Slater(HFS)方法计算COT2M(M=Th,Pa,U,Pu)结构中金属与环的键距离分别为0.208、0.202、0.198、0.197和0.196 nm[32]。采用多组态自洽场MCSCF方法计算COT2U得到铀与环的键距离为0.207 nm[33]。Moritiz和Dlog[18]采用将5f电子冻芯的赝势energy-consistent PPs和核极化赝势CPPs研究了COT2An(An=Th-Pu)的结构和结合能等性质,并比较MP2,CCDC以及密度泛函理论B3LYP和PW91不同方法优化得到的结构参数。该计算采用的赝势将5f电子当做内层电子进行冻芯处理,并且B3LYP计算得到铀与环的距离为0.202 5 nm[18]。Kerridge等[19]也通过CASPT2,在固定环结构参数情况下局部优化COT2U(和COT2Pu(结构得到金属到COT环中心的距离分别为0.194 4和0.189 8 nm。以上方法得到COT2U的铀与环的键距离都比实验值[34-35]0.192 4 nm大,并且都只针对气相下的结构进行计算,并未考虑溶剂化效应。理论上对Cp4U的研究主要是在20世纪70年代和80年代时期,采用的方法多为半经验方法和早期的密度泛函理论[36]。随后也很少通过更现代的方法进行探讨。
本文应用密度泛函理论的B3LYP方法,通过采用不同的有效核势(ECP)计算研究环辛四烯(COT2-)和环戊二烯(Cp-)的金属铀有机配合物(Cp4U和COT2U)和具有类似结构的钚金属有机化合物(Cp4Pu和COT2Pu)在气相和四氢呋喃溶剂中的结构和化学性质。
1 计算方法
标量相对论效应方法主要是在非相对论量子化学的波函数的哈密顿算符中增加考虑相对论mass-velosity和Darwin两项[36]。包含相对论效应计算的高斯软件主要是通过有效核势(ECP)考虑相对论。用标量相对论计算含铀体系的基态性质是行之有效的手段。本文采用Gaussian09软件[37]的B3LYP泛函[38-39]计算铀体系的基态性质。EMSL数据库[40]中有CRENBL ECP,“大核芯”有效赝势(RLC ECP)和“小核芯”有效赝势(RSC ECP)的赝势基组[41-43]。对U和Pu采用3种不同的赝势基组,其它原子采用6-31+g(d)全电子基组进行结构优化。CRENBL ECP和RLC ECP对U和Pu外层14或16个电子当成价电子处理,把内层78个电子冻芯处理。RSC-ECP则对对U和Pu外层32或34个电子当成价电子处理,把内层60个电子冻芯处理。计算中溶剂化效应采用CPCM模型。采用联合原子拓扑模型(UATM)构造空穴,原子半径采用UA0(simple united atom topological model)。四氢呋喃(THF)的介电常数采用ε=7.42。
2 结果与讨论
2.1 COT2An和Cp4An(An=U,Pu)基态的确定
如图 1,环辛四烯(COT2-)和环戊二烯(Cp-)的金属铀和钚有机配合物的结构中金属与配体成键形成夹心结构。Hayes和Edelstein[44]通过基础分子轨道计算预测COT2U和COT2Pu的基态结构得到|Jz|=4,0,并认为COT2U是磁性化合物而COT2Pu为反磁性化合物。为确定化合物的基态,计算考虑了多种自旋态的优化,结果如表 1所示。COT2U和COT2Pu配合物中金属的化合价为+4价,因为铀离子电子结构有2个5f电子,而钚离子电子结构中有4个5f电子。因此计算分别考虑了单重态、三重态的COT2U和单重态、三重态、五重态的COT2Pu。根据计算得到的体系能量大小可知,三重态COT2U和五重态COT2Pu属于基态结构。能量上,其它态的体系能量比基态体系能量高出140~500 kJ·mol-1。Kerridge等[19]也通过CASPT2优化金属-COT环中心的距离计算表明旋-轨耦合对COT2U和COT2Pu结构影响很小。由于COT2Pu的理论计算表明基态为五重态,而实际上却是反磁性物质的特征,在理论研究中也倍受争议[26, 44]。因此在计算中有必要考虑单重态的COT2Pu的结构。
图1
环辛四烯(COT2-)和环戊二烯(Cp-)的金属铀和钚有机化合物的结构(Cp4An和COT2An)
Figure1.
Structures of cyclopentadienyl (Cp-) and annulene(COT2-) sandwiched uranium and plutonium compounds (Cp4An and COT2An)
 表 1
气相下B3LYP/CRENBL ECP方法计算不同自旋态的COT2An和Cp4An(An=U(Ⅳ), Pu(Ⅳ))结构的优化结果(nm)和相对能量(kJ·mol-1)
Table 1.
Optimized geometry parameters (nm) and relative energies (kJ·mol-1) of COT2An and Cp4An with An=U(Ⅳ), Pu(Ⅳ) for different spin states by B3LYP/CRENBL ECP in gas phase
表 1
气相下B3LYP/CRENBL ECP方法计算不同自旋态的COT2An和Cp4An(An=U(Ⅳ), Pu(Ⅳ))结构的优化结果(nm)和相对能量(kJ·mol-1)
Table 1.
Optimized geometry parameters (nm) and relative energies (kJ·mol-1) of COT2An and Cp4An with An=U(Ⅳ), Pu(Ⅳ) for different spin states by B3LYP/CRENBL ECP in gas phase
Spin state RAn-ligand RC-C RAn-C Relative energy Singlet-COT2U 0.201 6 0.141 8 0.274 2 296.7 Triplet-COT2U 0.206 0 0.141 8 0.277 2 0 COT2U Exp.[34-35] 0.192 4 0.139 2 0.264 7 — SingletCOT2Pu 0.202 9 0.141 6 0.275 0 396.2 Triplet-COT2Pu 0.209 9 0.141 7 0.280 2 198.0 Quintet COT2Pu 0.209 3 0.141 8 0.279 4 0 Singlet-Cp4U 0.265 1 0.142 2 0.291 5 220.3 Triplet-Cp4U 0.270 4 0.142 4 0.298 5 0 Cp4UExp.[30] 0.253 8 0.138 6 0.280 7 — Singlet-Cp4Pu 0.264 2 0.142 3 0.290 0 469.5 Triplet-Cp4Pu 0.267 8 0.142 4 0.295 1 149.1 Quintet-Cp4Pu 0.266 7 0.142 3 0.293 2 0 表 1中,与相关的实验数据比较,三重态的COT2U和Cp4U中U-ligand的距离比实验值分别长0.013 6和0.016 6 nm。低自旋态的结构参数却与实验值更为接近,这可能是由于CRENBL ECP赝势高估了键长。因此有必要进行其它赝势基组计算。
2.2 不同的相对论赝势基组对几何结构的影响
表 2中CRENBL ECP,RLC ECP和RSC ECP赝势基组的优化结果存在一定程度的差异。CRENBL和RLC ECPs计算得到的键长比RSC ECP计算的键长结果长。对含铀和钚的体系采用小核芯的RSC ECP赝势基组可以得到与实验值[34-35]更为接近的结果。含钚体系中CRENBL和RLC ECPs计算得到的结构参数中键长比RSC ECP的计算结果约长0.01~0.02 nm。采用RSC ECP赝势基组计算得到COT2U中金属到环中心距离U-ligand为0.194 3 nm与实验数据0.192 4 nm一致[34-35]。同时采用RSC ECP赝势基组计算得到Cp4U中金属到环中心距离U-ligand为0.260 4 nm只比实验值[30]长0.007 6 nm。关于COT2Pu确切的结构参数未见实验报道,但有报道通过已知的锕系金属环状有机配合物的键长结果推算出Pu-ligand的数值为0.183 nm[19, 45]。不同自旋态COT2Pu的结构参数如表 2所示,采用RSC ECP赝势基组得到相对稳定的五重态的Pu-ligand的数值为0.197 4 nm,而不稳定的单重态的Pu-ligand的数值为0.193 4 nm。
表 2
气相和THF液相下不同赝势基组计算COT2An(和Cp4An (An=U(Ⅳ), Pu(Ⅳ))基态结构的优化结果(nm)
Table 2.
Optimized geometry parameters (nm) for ground states of COT2Anand Cp4An with An=U(Ⅳ), Pu(Ⅳ) by different ECPs in gas and THF solution phase
Species ECP Typ RAn-ligand RAn-C Gas THF Gas THF COT2U CRENBL 0.206 0 0.213 7 0.277 2 0.283 1 RLC 0.199 4 0.213 3 0.272 4 0.282 7 RSC 0.194 3 0.203 5 0.270 6 0.275 2 Exp.[34-35] 0.192 4 0.264 7 COT2Pu CRENBL 0.209 3 0.215 8 0.279 4 0.284 4 Quintet RLC 0.219 6 0.230 4 0.287 4 0.295 6 RSC 0.197 4 0.220 3 0.270 9 0.274 4 COT2Pu CRENBL 0.202 9 0.230 8 0.275 0 0.275 3 Singlet RLC 0.207 9 0.214 4 0.278 6 0.283 5 RSC 0.193 4 0.193 2 0.267 9 0.267 8 Cp4U CRENBL 0.270 4 0.273 3 0.298 5 0.298 8 RLC 0.265 7 0.267 4 0.291 8 0.292 8 RSC 0.260 4 0.260 8 0.286 5 0.287 4 Exp.[30] 0.253 8 0.280 7 Cp4Pu CRENBL 0.266 7 0.267 5 0.293 2 0.294 9 Quintet RLC 0.273 0 0.274 8 0.297 4 0.296 1 RSC 0.259 4 0.259 5 0.285 9 0.286 1 Cp4Pu CRENBL 0.264 2 0.265 2 0.290 0 0.290 0 Singlet RLC 0.267 2 0.271 0 0.292 5 0.296 1 RSC 0.258 0 0.257 8 0.283 8 0.284 3 在THF溶液中,An-ligand和An-C键受到溶剂化效应的影响都比气相中的结果长。其中COT2U的U-ligand增长了0.009 2 nm,五重态COT2Pu的Pu-ligand增长了0.022 9 nm,但是单重态COT2Pu在两相中相应键长相似。环戊二烯化合物Cp4U(和Cp4Pu的结构参数受THF溶剂的影响也很小,但溶剂化效应对COT2U和COT2Pu的影响很大。Berthet等[46]合成了弯曲的环辛四烯铀的衍生物说明,被2个COT2-环夹心的铀在溶液中可能受到环境中其它配体的作用而使两环成一定夹角而弯曲。
2.3 结合能计算
COT2An和Cp4An(An=U(Ⅳ),Pu(Ⅳ))中金属与环之间的结合能如表 3所示。结合能采用下面的公式计算:
表 3
气相和THF液相下不同赝势基组计算得到的COT2An和Cp4An (An=U(Ⅳ), Pu(Ⅳ))结合能(eV)
Table 3.
Actinide-ring binding energies (eV) for Cp4An and COT2An with An=U(Ⅳ), Pu(Ⅳ) by different ECPs in gas and THF solution phase
Species CRENBL ECP RLC ECP RSC ECP Eb-Gas Eb-THF Eb-Gas Eb-THF Eb-Gas Eb-THF COT2U -93.8 -25.9 -92.2 -24.1 -90.7 -23.7 COT2Pu(Quintet) -95.0 -29.0 -94.5 -28.4 -95.9 -28.2 COT2Pu(Singlet) -97.5 -28.4 -96.6 -28.3 -96.3 -27.4 Cp4U -86.2 -23.9 -86.1 -21.9 -84.2 -21.9 Cp4Pu(Quintet) -86.8 -26.7 -86.5 -26.2 -88.4 -26.6 Cp4Pu(Singlet) -88.5 -25.6 -87.8 -25.3 -87.7 -24.9 其中Eb、Ecomplex、Eactinide ion和Ecyclo-ligand分别表示结合能、金属有机化合物体系的能量、锕系金属离子的能量和环状有机配体的能量。所有能量值都通过零点能(ZPE)校正。n表示环状有机配体的数目。
表 3中所采用的不同ECPs赝势基组都表明COT2An和Cp4An(An=U(Ⅳ),Pu(Ⅳ))中金属与环之间的结合能很大,进而说明在气相和THF液相中环状有机配体与锕系金属离子之间有很强的成键作用。含COT2-配合物的结合能比相应的含Cp-配合物的能量高。Moritiz和Dlog[18]研究发现气相中COT2An锕系金属有机配合物的结合能随中心金属核电荷数的增加而增大。表 3中含钚的金属有机配合物也比相应的含铀有机配合物的结合能大。CRENBL和RLC ECPs计算的结果表明,气相中金属钚有机配合物比类似结构的金属铀有机配合物稳定,其结合能大1 eV左右。金属钚有机配合物的稳定性在THF溶液中尤为突出,其结合能比类似结构的金属铀配合物大4 eV左右。但是采用RSC-ECP的计算却表明不管在气相还是THF溶剂中金属钚有机配合物的结合能比类似结构的金属铀有机配合物的结合能大4 eV。溶剂化效应使得配合物的结合能相对气相中的结合能下降了接近70%。实验上合成得到的红色晶体状的Cp4U[28]在空气中能够稳定存在,但在苯溶液中的结构却不稳定,也说明了溶剂化对结构稳定性的影响很大。从计算给出的结果可以说明,该类化合物在气相当中比较稳定,保存的环境考虑为气相。使用不同的相对论赝势对结合能的数值影响比较大。气相中RSC ECP的结果显示COT2Pu五重态的结合能为95.9 eV,而其单重态的结合能为96.3 eV。而考虑THF溶剂化效应后,COT2Pu五重态的结合能反而比其单重态的结合能高。Cp4Pu配合物的结合能也有同样的趋势。这说明溶剂可以使Cp4Pu和COT2Pu的基态发生变化。
2.4 红外光谱性质
采用最佳的RSC ECP赝势基组得到的红外光谱模拟图如图 2所示。通过计算得到气相中的COT2U的红外吸收锋有724、923 cm-1(medium)和3 183 cm-1(strong);THF液相中其红外吸收峰有712 cm-1(medium)、918 cm-1(medium)和3 177 cm-1(medium)。而在气相和THF液相中COT2U的理论红外吸收分别在1 556 cm-1(strong)和1 518 cm-1(strong)处有峰,该峰是C8H8环中C-H的上下振动频率。气相中COT2U、Cp4U、COT2Pu和Cp4Pu分别在724 cm-1(strong)、788 cm-1(medium)、702 cm-1(strong)和772 cm-1处的红外吸收光谱属于有机环的伸缩振动特征光谱。而结构中环中C-H键红外特征吸收光谱出现在1 000~1 500 cm-1波数范围,同时在THF溶剂中配合物的红外光谱吸收峰具有更高的强度。由此可见,理论计算预算得到的环辛四烯(COT2-)和环戊二烯(Cp-)配体在在800 cm-1波数以下的指纹区域有强的有机环的伸缩振动特征吸收峰。实验测得COT2U的红外光谱吸收峰有:594 cm-1(strong)、741 cm-1(medium)、772 cm-1、787 cm-1、900 cm-1(medium)和3 000 cm-1 [47]。结果表明理论红外光谱计算结果与实验侧得的数据吻合,也进一步说明该化合物的结构优化结果合理。
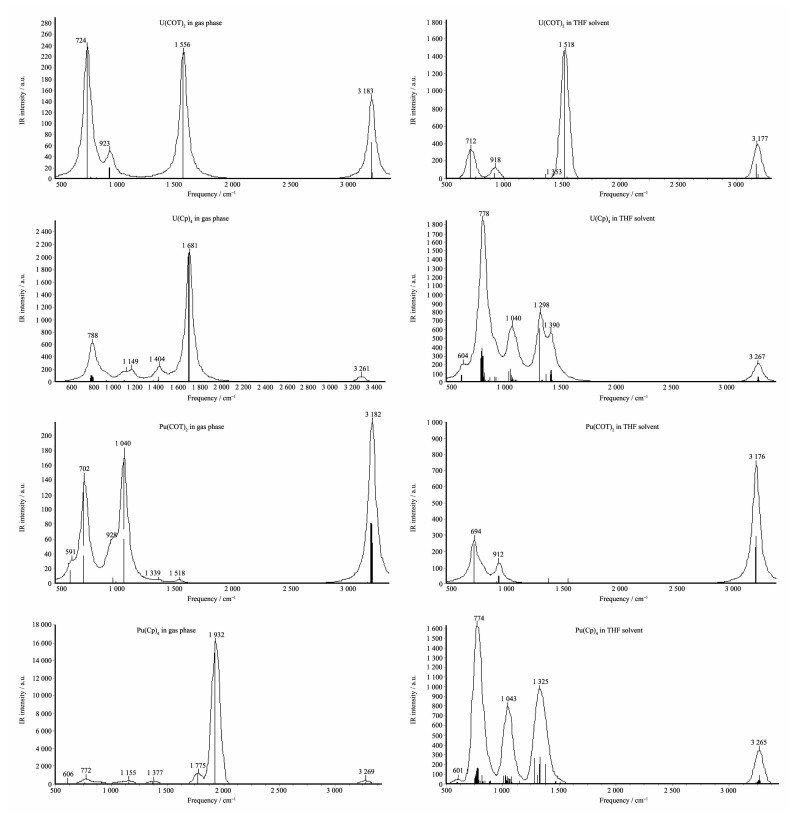 图2
气相和THF液相下COT2U, COT2Pu, Cp4U和Cp4Pu的红外光谱模拟
Figure2.
IR absorption spectra of COT2U, COT2Pu, Cp4U, and Cp4Pu in gas phase and THF solution
图2
气相和THF液相下COT2U, COT2Pu, Cp4U和Cp4Pu的红外光谱模拟
Figure2.
IR absorption spectra of COT2U, COT2Pu, Cp4U, and Cp4Pu in gas phase and THF solution
2.5 分子轨道
RSC ECP计算得到An(Cp)4和An(COT)2(An= U(Ⅳ),Pu(Ⅳ))的分子级轨道能级图(图 3)中能量低于-8 eV能级的电子受自旋影响小,可明显观测到能量相等α电子与β电子配对存在于分子轨道能级中。由于锕系元素电子排布的复杂性,很难准确确定其基态电子结构。通过采用RSC赝势的计算进行预测,配合物An(COT)2的分子轨道能级图显示含5fφ电子的轨道不与COT环的轨道作用,5f电子主要分布在能量范围在-4~-8 eV区域的最高占据轨道上。配合物U(COT)2的H-0和H-1轨道属于5fφ电子轨道,而配合物Pu(COT)2中H-3,H-4,H-5,H-6轨道属于5fφ,5fφ,5fπ和5fσ电子轨道。由此可见U4+和Pu4+离子中5f电子在形成配合物时部分5f电子不与环配体电子作用,而单独占据电子轨道。含Pu有机配合物的结合能大小可能与轨道中的5f单电子有关。同时从图 3中也可以清晰地看到配合物An(COT)2的成键轨道H-2,H-3(U(COT)2)和H-0(Pu(COT)2)存在环配体与金属5f电子相互作用形成的成键轨道。该成键轨道是通过配体提供电子对,金属供空的fδ轨道而形成的。与2个COT环形成成键轨道还包含了部分空的6dδ轨道,比如H-4(U(COT)2)和H-1,H-2(Pu(COT)2)轨道。金属U或Pu与COT环的成键作用,比其与Cp环的成键作用强。根据图 3的分子轨道能级图可判断U(COT)2是fφ2组成的三重态结构,而Pu(COT)2则是fφ2fπ1fσ1组成的五重态结构。King[48]应用拓扑轨道理论预测An(Cp)4和An(COT)2(An=Th,Pa,U,Np)金属有机化合物的5f价电子首先占据5fφ轨道。本章节对U(COT)2的轨道能级计算与King的结论一致[48]。但对于Pu(COT)2化合物f电子只有2个电子占据5fφ轨道。由于Pu电子结构的复杂性,其配合物的基态结构还有待实验上进一步验证。配合物An(Cp)4中,配位键主要是由5fφ空轨道与Cp环的2p轨道作用形成。U(Cp)4是三重态的fσ2结构,Pu(Cp)4则为五重态的fσ3fδ1结构。
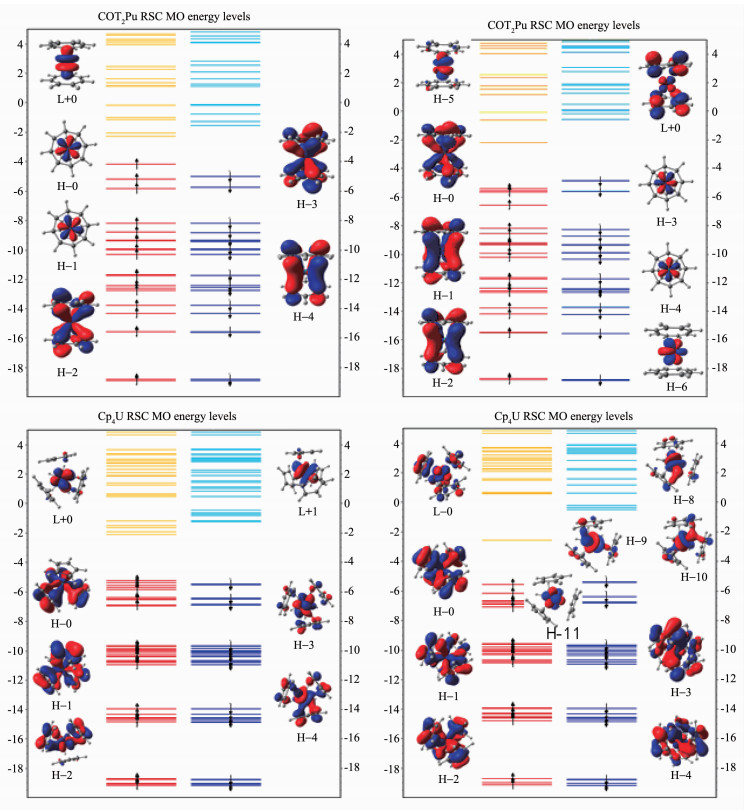 图3
RSC ECP计算得到的An(Cp)4和An(COT)2(An=U(Ⅳ), Pu(Ⅳ))分子轨道能级图(部分前线轨道图显示在能级图上, 其中H代表HOMO轨道, L代表LUMO轨道)
Figure3.
MO energy level diagrams for An(Cp)4 and An(COT)2 with An=U(Ⅳ), Pu(Ⅳ) calculated with RSC ECP (Partial frontier orbital are shown in energy level diagrams with H and L shorted for the highest occupied and lowest unoccupied orbital respectively)
图3
RSC ECP计算得到的An(Cp)4和An(COT)2(An=U(Ⅳ), Pu(Ⅳ))分子轨道能级图(部分前线轨道图显示在能级图上, 其中H代表HOMO轨道, L代表LUMO轨道)
Figure3.
MO energy level diagrams for An(Cp)4 and An(COT)2 with An=U(Ⅳ), Pu(Ⅳ) calculated with RSC ECP (Partial frontier orbital are shown in energy level diagrams with H and L shorted for the highest occupied and lowest unoccupied orbital respectively)
3 结论
本文采用Gaussian09中的密度泛函中的B3LYP泛函研究配合物COT2U、COT2Pu、Cp4U和Cp4Pu的几何结构和电子结构。优化结果表明对含铀和含钚的体系进行计算需采用小核冻芯的RSC相对论有效赝势。计算得到该类有机物的IR光谱与实验值一致,其中红外特征吸收峰在500~1 000 cm-1区域。结合能计算表明配合物的稳定性呈COT2Pu>COT2U>Cp4Pu>Cp4U的趋势,因此含U和Pu的金属有机配合物主要是通过配位键而稳定存在的。配位键主要是由5f空轨道与COT或Cp环的2p占据轨道作用形成。分子轨道能级图可判断U(COT)2是fφ2组成的三重态结构,而Pu(COT)2则是fφ2fπ1fσ1组成的五重态结构,U(Cp)4是三重态的fσ2结构,Pu(Cp)4则为五重态的fσ3fδ1结构。
-
-
[1]
Clark D L, Hobart D E, Neu M P. Chem. Rev., 1995, 95(1):25-48 doi: 10.1021/cr00033a002
-
[2]
Craw J S, Vincent M A, Hillier I H. J. Phys. Chem. A, 1995, 99:10181-10185 doi: 10.1021/j100025a019
-
[3]
Hemmingsen L, Amara P, Ansoborlo E, et al. J. Phys. Chem. A, 2000, 104(17):4095-4101 doi: 10.1021/jp994395o
-
[4]
Privalov T, Schimmelpfennig B, Wahlgren U, et al. J. Phys. Chem. A, 2002, 106(46):11277-11282 doi: 10.1021/jp0260402
-
[5]
de Jong W A, Aprà E, Windus T L, et al. J. Phys. Chem. A, 2005, 109(50):11568-11577 doi: 10.1021/jp0541462
-
[6]
Denecke M A. Coord. Chem. Rev., 2006, 250(7/8):730-754
-
[7]
辜家芳, 陆春海, 陈文凯, 等.物理化学学报, 2009, 25(4):655-660 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmGU Jia-Fang, LU Chun-Hai, CHEN Wen-Kai, et al. Acta Phys.-Chim. Sin., 2009, 25(4):655-660 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[8]
辜家芳, 陆春海, 陈文凯, 等.物理化学学报, 2012, 28(4):792-798 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmGU Jia-Fang, LU Chun-Hai, CHEN Wen-Kai, et al. Acta Phys.-Chim. Sin., 2012, 28(4):792-798 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[9]
辜家芳, 满梅玲, 陆春海, 等.无机化学学报, 2012, 28(7):1324-1332 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20120704&journal_id=wjhxxbcnGU Jia-Fang, MAN Mei-Ling, LU Chun-Hai, et al. Chinese J. Inorg. Chem., 2012, 28(7):1324-1332 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20120704&journal_id=wjhxxbcn
-
[10]
Tatsumi K, Nakamura A. J. Organomet. Chem., 1984, 272(2):141-154 doi: 10.1016/0022-328X(84)80462-3
-
[11]
Tatsumi K, Nakamura A. J. Am. Chem. Soc., 1987, 109(11):3195-3206 doi: 10.1021/ja00245a003
-
[12]
Chang A H H, Pitzer R M. J. Am. Chem. Soc., 1989, 111(7):2500-2507 doi: 10.1021/ja00189a022
-
[13]
Gourier D, Caurant D, Berthet J C, et al. Inorg. Chem., 1997, 36(25):5931-5936 doi: 10.1021/ic961496h
-
[14]
Li J, Bruce E B. J. Am. Chem. Soc., 1997, 119:9021-9032 doi: 10.1021/ja971149m
-
[15]
Li J, Bursten B E. J. Am. Chem. Soc., 1998, 120(44):11456-11466 doi: 10.1021/ja9821145
-
[16]
Hager J S, Zahardis J, Pagni R M, et al. J. Chem. Phys., 2004, 120(6):2708-2718 doi: 10.1063/1.1637586
-
[17]
BenYahia M, Belkhiri L, Boucekkine A. J. Mol. Struct., 2006, 777(1/2/3):61-73
-
[18]
Moritz A, Dolg M. Chem. Phys., 2007, 337(1/2/3):48-54
-
[19]
Kerridge A, Kaltsoyannis N. J. Phys. Chem. A, 2009, 113(30):8737-8745 doi: 10.1021/jp903912q
-
[20]
Streitwieser A. J. Org. Chem., 2009, 74(12):4433-4446 doi: 10.1021/jo900497s
-
[21]
Elkechai A, Meskaldji S, Boucekkine A, et al. J. Mol. Struct., 2010, 954(1/2/3):115-123
-
[22]
Yahia A, Castro L, Maron L. Chem. Eur. J., 2010, 16(19):5564-5567 doi: 10.1002/chem.201000022
-
[23]
Zhou J, Sonnenberg J L, Schlegel H B. Inorg. Chem., 2010, 49(14):6545-6551 doi: 10.1021/ic100427t
-
[24]
Castro L, Yahia A, Maron L. C. R. Chim., 2010, 13(6/7):870-875
-
[25]
Streitwieser A, Mueller-Westerhoff U. J. Am. Chem. Soc., 1968, 90(26):7364-7364
-
[26]
Karraker D G, Stone J A, Jones E R, et al. J. Am. Chem. Soc., 1970, 92(16):4841-4845 doi: 10.1021/ja00719a014
-
[27]
Streitwieser J A, Muller-Westerhoff U, Sonnichsen G, et al. J. Am. Chem. Soc., 1972, 95(26):8644-8649
-
[28]
Fischer V E O, Hristidu Y. Z. Naturforsch., 1962, 17b:275-276
-
[29]
Burns J H. J. Organomet. Chem., 1974, 69(2):225-233 doi: 10.1016/S0022-328X(00)90241-9
-
[30]
Burns J H. J. Am. Chem. Soc., 1973, 95:3815-3817 doi: 10.1021/ja00792a070
-
[31]
Kanellakopulos B, Maier R, Heuser J. J. Alloys Compd., 1991, 176(1):89-96 doi: 10.1016/0925-8388(91)90013-L
-
[32]
Boerrigter P M, Baerends E J, Snijders J G. Chem. Phys., 1988, 122(3):357-374 doi: 10.1016/0301-0104(88)80018-1
-
[33]
Liu W, Dolg M, Fulde P. J. Chem. Phys., 1997, 107(9):3584-3591 doi: 10.1063/1.474698
-
[34]
Zalkin A, Raymond K N. J. Am. Chem. Soc., 1969, 91:5667-5668 doi: 10.1021/ja01048a055
-
[35]
Avdeef A, Raymond K N, Hodgson K O, et al. Inorg. Chem., 1972, 11:1083-1088 doi: 10.1021/ic50111a034
-
[36]
Kaltsoyannis N, Hay P J, Li J, et al. Chemistry of the Actinide and Transactinide Elements. Morss L R, Edelstein N M, Fuger J Eds., Netherlands:Springer, 2006:1893-2012
-
[37]
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, Wallingford CT:Gaussian, Inc., 2009.
-
[38]
Becke A D. Phys. Rev. A, 1988, 38:3098-3100 doi: 10.1103/PhysRevA.38.3098
-
[39]
Bardin N, Rubini P, Madic C. Radiochim. Acta, 1998, 83:189-194
-
[40]
Gaussian Basis Set Order Form:http://www.emsl.pnl.gov/forms/basisform.html
-
[41]
Hay P J. J. Chem. Phys., 1983, 79:5469-5482 doi: 10.1063/1.445665
-
[42]
Kahn L R, Hay P J, Cowan R D. J. Chem. Phys., 1978, 68:2386-2397 doi: 10.1063/1.436009
-
[43]
Feller D. J. Comput. Chem., 1996, 17(13):1571-1586 doi: 10.1002/(ISSN)1096-987X
-
[44]
Hays R G, Edelstein N M. J. Am. Chem. Soc., 1972, 94(25):8688-8691 doi: 10.1021/ja00780a008
-
[45]
Shannon R. Acta Crystallogr. Sect. A, 1976, 32(5):751-767 doi: 10.1107/S0567739476001551
-
[46]
Berthet J C, Thuéry P, Ephritikhine M. Organometallics, 2008, 27(8):1664-1666 doi: 10.1021/om800006u
-
[47]
Weakliem P C, Carter E A. J. Chem. Phys., 1992, 96:3240-3250 doi: 10.1063/1.461968
-
[48]
King R B. Inorg. Chem., 1992, 31(10):1978-1980 doi: 10.1021/ic00036a052
-
[1]
-

图 1 环辛四烯(COT2-)和环戊二烯(Cp-)的金属铀和钚有机化合物的结构(Cp4An和COT2An)
Figure 1 Structures of cyclopentadienyl (Cp-) and annulene(COT2-) sandwiched uranium and plutonium compounds (Cp4An and COT2An)

图 2 气相和THF液相下COT2U, COT2Pu, Cp4U和Cp4Pu的红外光谱模拟
Figure 2 IR absorption spectra of COT2U, COT2Pu, Cp4U, and Cp4Pu in gas phase and THF solution

图 3 RSC ECP计算得到的An(Cp)4和An(COT)2(An=U(Ⅳ), Pu(Ⅳ))分子轨道能级图(部分前线轨道图显示在能级图上, 其中H代表HOMO轨道, L代表LUMO轨道)
Figure 3 MO energy level diagrams for An(Cp)4 and An(COT)2 with An=U(Ⅳ), Pu(Ⅳ) calculated with RSC ECP (Partial frontier orbital are shown in energy level diagrams with H and L shorted for the highest occupied and lowest unoccupied orbital respectively)
表 1 气相下B3LYP/CRENBL ECP方法计算不同自旋态的COT2An和Cp4An(An=U(Ⅳ), Pu(Ⅳ))结构的优化结果(nm)和相对能量(kJ·mol-1)
Table 1. Optimized geometry parameters (nm) and relative energies (kJ·mol-1) of COT2An and Cp4An with An=U(Ⅳ), Pu(Ⅳ) for different spin states by B3LYP/CRENBL ECP in gas phase
Spin state RAn-ligand RC-C RAn-C Relative energy Singlet-COT2U 0.201 6 0.141 8 0.274 2 296.7 Triplet-COT2U 0.206 0 0.141 8 0.277 2 0 COT2U Exp.[34-35] 0.192 4 0.139 2 0.264 7 — SingletCOT2Pu 0.202 9 0.141 6 0.275 0 396.2 Triplet-COT2Pu 0.209 9 0.141 7 0.280 2 198.0 Quintet COT2Pu 0.209 3 0.141 8 0.279 4 0 Singlet-Cp4U 0.265 1 0.142 2 0.291 5 220.3 Triplet-Cp4U 0.270 4 0.142 4 0.298 5 0 Cp4UExp.[30] 0.253 8 0.138 6 0.280 7 — Singlet-Cp4Pu 0.264 2 0.142 3 0.290 0 469.5 Triplet-Cp4Pu 0.267 8 0.142 4 0.295 1 149.1 Quintet-Cp4Pu 0.266 7 0.142 3 0.293 2 0  下载: 导出CSV
下载: 导出CSV
表 2 气相和THF液相下不同赝势基组计算COT2An(和Cp4An (An=U(Ⅳ), Pu(Ⅳ))基态结构的优化结果(nm)
Table 2. Optimized geometry parameters (nm) for ground states of COT2Anand Cp4An with An=U(Ⅳ), Pu(Ⅳ) by different ECPs in gas and THF solution phase
Species ECP Typ RAn-ligand RAn-C Gas THF Gas THF COT2U CRENBL 0.206 0 0.213 7 0.277 2 0.283 1 RLC 0.199 4 0.213 3 0.272 4 0.282 7 RSC 0.194 3 0.203 5 0.270 6 0.275 2 Exp.[34-35] 0.192 4 0.264 7 COT2Pu CRENBL 0.209 3 0.215 8 0.279 4 0.284 4 Quintet RLC 0.219 6 0.230 4 0.287 4 0.295 6 RSC 0.197 4 0.220 3 0.270 9 0.274 4 COT2Pu CRENBL 0.202 9 0.230 8 0.275 0 0.275 3 Singlet RLC 0.207 9 0.214 4 0.278 6 0.283 5 RSC 0.193 4 0.193 2 0.267 9 0.267 8 Cp4U CRENBL 0.270 4 0.273 3 0.298 5 0.298 8 RLC 0.265 7 0.267 4 0.291 8 0.292 8 RSC 0.260 4 0.260 8 0.286 5 0.287 4 Exp.[30] 0.253 8 0.280 7 Cp4Pu CRENBL 0.266 7 0.267 5 0.293 2 0.294 9 Quintet RLC 0.273 0 0.274 8 0.297 4 0.296 1 RSC 0.259 4 0.259 5 0.285 9 0.286 1 Cp4Pu CRENBL 0.264 2 0.265 2 0.290 0 0.290 0 Singlet RLC 0.267 2 0.271 0 0.292 5 0.296 1 RSC 0.258 0 0.257 8 0.283 8 0.284 3
下载: 导出CSV
表 3 气相和THF液相下不同赝势基组计算得到的COT2An和Cp4An (An=U(Ⅳ), Pu(Ⅳ))结合能(eV)
Table 3. Actinide-ring binding energies (eV) for Cp4An and COT2An with An=U(Ⅳ), Pu(Ⅳ) by different ECPs in gas and THF solution phase
Species CRENBL ECP RLC ECP RSC ECP Eb-Gas Eb-THF Eb-Gas Eb-THF Eb-Gas Eb-THF COT2U -93.8 -25.9 -92.2 -24.1 -90.7 -23.7 COT2Pu(Quintet) -95.0 -29.0 -94.5 -28.4 -95.9 -28.2 COT2Pu(Singlet) -97.5 -28.4 -96.6 -28.3 -96.3 -27.4 Cp4U -86.2 -23.9 -86.1 -21.9 -84.2 -21.9 Cp4Pu(Quintet) -86.8 -26.7 -86.5 -26.2 -88.4 -26.6 Cp4Pu(Singlet) -88.5 -25.6 -87.8 -25.3 -87.7 -24.9
下载: 导出CSV
-




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 1
- 文章访问数: 1286
- HTML全文浏览量: 182
 下载:
下载:
