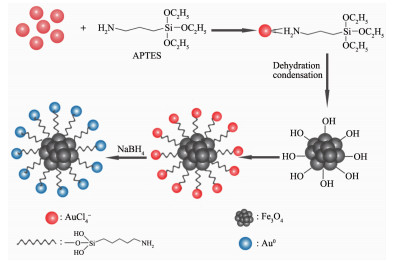
 图 Scheme 1
Process of synthesis of Au/ Fe3O4
图 Scheme 1
Process of synthesis of Au/ Fe3O4
引用本文:
顾岩, 刘冠廷, 邹铖亨, 张真, 刘娟, 孙明, 程高, 余林. 超声法制备Au/Fe3O4及其催化还原4-硝基苯酚性能[J]. 无机化学学报,
2017, 33(5): 787-795.
doi:
10.11862/CJIC.2017.102

Citation: GU Yan, LIU Guan-Ting, ZOU Cheng-Heng, ZHANG Zhen, LIU Juan, SUN Ming, CHENG Gao, YU Lin. Ultrasonic Synthesis and Application in Catalytic 4-Nitrophenols Reduction of Au/Fe3O4[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(5): 787-795. doi: 10.11862/CJIC.2017.102
Citation: GU Yan, LIU Guan-Ting, ZOU Cheng-Heng, ZHANG Zhen, LIU Juan, SUN Ming, CHENG Gao, YU Lin. Ultrasonic Synthesis and Application in Catalytic 4-Nitrophenols Reduction of Au/Fe3O4[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(5): 787-795. doi: 10.11862/CJIC.2017.102
超声法制备Au/Fe3O4及其催化还原4-硝基苯酚性能
摘要:
通过简易的超声法以及原位还原法成功制备出了负载型可再生Au/Fe3O4催化剂。利用3-氨丙基三乙氧基硅烷(APTES)作为有机桥键,将Au固定在Fe3O4的表面,得到单分散磁性Au/Fe3O4。Au0在氨基的作用下不会团聚,因此具有较高的催化活性及稳定性。XRD、HRTEM、EDS和XPS等测试结果表明Au/Fe3O4已被成功制备。将其用于催化还原4-硝基苯酚得到4-氨基苯酚,表现出较高的催化活性,速率常数可达0.225 6 min-1。重复性实验表明该催化剂具有良好的稳定性,反应9个循环之后,催化还原反应的转化率仍可达到94%。
English
Ultrasonic Synthesis and Application in Catalytic 4-Nitrophenols Reduction of Au/Fe3O4
Abstract:
The Au/Fe3O4 catalyst was successfully prepared by the simple ultrasonic method and in-situ reduction method. Au was immobilized on the surface of Fe3O4 using APTES as an organic bridging bond to obtain monodisperse magnetic Au/Fe3O4. Au0 could not agglomerated in the condition of NH3+, so that it had high catalytic activity and stability. The results of XRD, HRTEM, EDS and XPS showed that Au/Fe3O4 has been prepared successfully. It was used to catalyze the reduction of 4-nitrophenols to 4-aminophenols, which showed high catalytic activity with rate constant of 0.225 6 min-1. The repeatability test showed that the catalyst had good stability. After 9 cycles of reaction, the conversion rate of the reaction was still 94%.
-
Key words:
- Au nanoparticles
- / Fe3O4
- / 4-nitrophenols
- / catalytic reduction
-
Haruta首次报道了负载型金纳米颗粒 (Au NPs) 对催化CO的低温氧化反应的超高活性[1],自此之后,便引起广大学者对Au催化剂的极大兴趣。随后,Zhang等[2]发现Au NPs的尺寸是决定其催化性能的关键因素。随着尺寸的减小,其催化剂活性越高,当尺寸下降到亚纳米簇甚至单原子的时候,Au NPs表现出超高的催化活性。此外,减小Au NPs的尺寸不仅可以提升其催化活性,还可以提高贵金属的利用率,有助于开发低成本的高效工业催化剂。
磁性纳米颗粒特别是磁铁矿 (Fe3O4) 具有独特的磁性,可作为负载贵金属催化剂的载体。Fe3O4在作为载体时可以有效防止贵金属团聚从而提高其催化活性及稳定性。相关研究结果表明,可使用SiO2[3-4]、石墨烯[5]、3-氨丙基三乙氧基硅烷 (3-Aminopropyltrie-thoxysilane,APTES)[6]或者聚合物[7]对Fe3O4的表面进行改性,通过引入氨基[8]、羧基等有机官能团,负载的贵金属更加稳定,使Fe3O4具有更广阔的应用前景。例如,Nie等[6]将共沉淀方法制备的Fe3O4纳米颗粒作为核,利用APTES将其表面氨基功能化,再通过超声降解来还原Au3+以形成金涂层,成功制备了Fe3O4@Au纳米颗粒。Qu等[9]制备了一种多功能纳米复合材料Fe3O4@mSiO2@Au,并应用于pH值响应和靶向药物传送。Rostamizade等[10]将Au锚定在硫醇官能化的超顺磁性Fe3O4纳米颗粒的表面,用作于炔烃水合反应的催化剂,显示出非常高的催化效率。Zhu等[11]采用聚醚酰亚胺作为配位剂和光还原剂,通过区域选择性光还原法制备了磁性可分离的Fe3O4@SiO2@PEI@Au催化剂,并将其用其催化还原4-硝基苯酚,显示出了良好的催化活性。
4-硝基苯酚 (4-nitrophenol,4-NPs) 在各种工业如印刷,纺织,石油化工和制药工业等的废水中广泛存在,该物质在水中具有高溶解性和稳定性,使之成为较难处理的水体污染物之一[12-13]。4-NPs不仅会导致严重的生态问题,还会对人类和野生动物造成致癌或遗传毒性等影响[14]。现已有多种处理技术[15-17],如Fenton反应、光催化及生物降解等,被应用于降解4-NPs。然而,这些方法都存在应用方面的局限性,例如苛刻的pH值、二次污染或者昂贵的后处理费等。据相关研究报道,用硼氢化钠还原4-NPs获得重要的工业中间体4-氨基苯酚 (4-aminophenol,4-APs) 被认为是处理4-NPs的有效方法[18-19]。该方法操作简单、安全无污染,且4-NPs可完全转化成4-APs,无副产物生成。但是,该反应需要加入催化剂才能进行,因此,研究可用于有效降解4-NPs的催化剂具有十分重要的意义。
本文采用一种简易的超声法以及原位还原法成功制备出Au/Fe3O4催化剂,合成路线如过程1所示。先利用APTES的氨基NH3+与氯金酸根离子AuCl4-的静电吸引形成APTES-Au;再通过APTES水解缩合将AuCl4-负载到Fe3O4表面;最后,通过硼氢化钠将Au3+原位还原成Au0从而得到单分散磁性Au/Fe3O4纳米微球。该方法较之前广泛采用的回流法而言,操作简单、绿色无污染且所需时间少。将Au/Fe3O4用于催化还原4-NPs,显示出良好的催化活性,反应完全仅需要8 min。
图 Scheme 1
Process of synthesis of Au/ Fe3O4
1 实验部分
1.1 主要试剂
柠檬酸钠、乙二醇、六水合三氯化铁 (FeCl3·6H2O)、尿素、3-氨丙基三乙氧基硅烷 (APTES)、氨水、硼氢化钠 (NaBH4) 购于阿拉丁试剂有限公司;氯金酸 (HAuCl4·3.5H2O) 购于上海利铂试剂有限公司;无水乙醇购于广州化学试剂厂。所有试剂未经处理,直接用于反应。
1.2 实验步骤
1.3 材料表征
X射线衍射 (X-ray diffraction,XRD,RIGAKU ULTIMA-Ⅲ) 使用Cu Kα射线 (λ=0.154 06 nm) 在5°~90°(2θ) 的范围扫描,管电压为40 kV,管电流为40 mA;使用扫描电子显微镜 (Scanning Electron Micros-cope,SEM,JSM-7001F,JEOL) 和高分辨率透射电子显微镜 (High Resolution Transmission Electron Micros-copy,HRTEM,FEI Tecnai G20) 表征样品的形态和尺寸,工作电压分别为15 kV和200 kV。采用能量分散X射线分析仪 (Energy Dispersive Spectrometer,EDS,Hitachi,Japan) 分析表面元素组成。通过X射线光电子能谱 (X-ray photoelectron spectroscopy, XPS, Thermo ESCALAB 250XI) 分析元素所处的化学环境。通过电感耦合等离子体-原子发射光谱法 (ICP-AES,Perkin Elmer Nexion 300) 分析元素含量。使用紫外-可见光分光光度计 (UNICO UV2800) 监测整个催化反应的进行。
1.4 性能测试
硼氢化钠还原4-NPs的反应在比色管 (25 mL) 中完成,即在室温下在比色管中加入3.5 mL 4-NPs溶液 (1 mmol·L-1),3.5 mL NaBH4溶液 (0.1 mol·L-1) 以及17 mL H2O。随后,向混合物中加入1 mL本文制备的Au/Fe3O4水溶液 (1 mg·mL-1),随着反应物的颜色由亮黄色逐渐变为无色,标志着反应的完成,整个反应在紫外-可见光分光光度计的监测下进行。
1.5 Au/Fe3O4的重复性测试
将催化反应后的催化剂在外加磁场的作用下富集起来,用蒸馏水洗涤3次后,重新分散在蒸馏水中,超声形成均匀的悬浮液 (1 mg·mL-1)。重复1.4步骤中的过程8次,并记录每一次的实验结果。
1.2.1 Fe3O4纳米颗粒的合成
称取0.32 g柠檬酸钠加入30 mL乙二醇中,在磁力搅拌和加热条件下使其完全溶解。待其冷却至室温,再加入0.97 g FeCl3·6H2O和0.9 g尿素,继续搅拌1 h以确保所有物质均已溶解。将上述溶液转移到100 mL的不锈钢反应釜中,置于烘箱,200 ℃反应12 h。反应结束后,取出反应产物,用磁铁分离并用无水乙醇和蒸馏水交替洗涤3次,并重新分散在无水乙醇 (0.25%质量百分数) 中备用。
1.2.2 Au/Fe3O4纳米颗粒的合成
取2 g Fe3O4悬浮液分散于100 g无水乙醇中,将溶液转至250 mL三口烧瓶中。机械搅拌并超声分散,加入2 mL去离子水以及0.6 mL氨水,持续搅拌形成A液。取0.8 g APTES分散至8 g无水乙醇中,再把1 mL 0.1 mg·mL-1氯金酸的乙醇溶液分散至8 g无水乙醇中,将氯金酸的乙醇溶液快速加入APTES的乙醇溶液,形成B液。在机械搅拌以及超声条件下将B液缓慢滴加至A液中。滴加完毕后,关超声并持续搅拌12 h。反应结束后,分别用无水乙醇和蒸馏水交替洗涤产物3次,用磁铁将黑色沉淀物分离后再分散至60 g去离子水中。最后再将溶液转至三口烧瓶中,机械搅拌辅助超声,加入5 mL NaBH4(0.1 mol·L-1),持续反应15 min后,取出产物,用去离子水洗涤3次,用磁铁将产物分离后放在真空干燥箱中60 ℃干燥12 h备用。
1.2.3 Au/Fe3O4-1纳米颗粒的合成
采用分步法,合成Au/Fe3O4-1。取2 g Fe3O4悬浮液分散于100 g无水乙醇中,将溶液转至250 mL三口烧瓶中。机械搅拌并超声分散,加入2 mL去离子水以及0.6 mL氨水,持续搅拌形成A液。取0.8 g APTES分散至10 g无水乙醇中,形成B液。在机械搅拌以及超声条件下将B液缓慢滴加至A液中。滴加完毕后,关超声并持续搅拌12 h。反应结束后,分别用无水乙醇和蒸馏水交替洗涤产物3次,用磁铁将黑色沉淀物分离后再分散至60 g去离子水中。然后将溶液转至三口烧瓶中,机械搅拌辅助超声,加入1 mL 0.1 mg·mL-1氯金酸的水溶液,持续反应30 min。最后再加入5 mL NaBH4(0.1 mol·L-1),持续反应15 min后,取出产物,用去离子水洗涤3次,用磁铁将产物分离后放在真空干燥箱中60 ℃干燥12 h备用。
1.2.4 Fe3O4-Au纳米颗粒的合成
通过“一锅煮”的方法直接合成Fe3O4-Au。称取0.32 g柠檬酸钠加入30 mL乙二醇中,在磁力搅拌和加热条件下使其完全溶解。待其冷却至室温,再加入0.97 g FeCl3·6H2O、1 mL 0.1 mg·mL-1氯金酸乙醇溶液和0.9 g尿素,继续搅拌1 h以确保所有物质均已溶解。将上述溶液转移到100 mL的不锈钢反应釜中,置于烘箱,200 ℃反应12 h。反应结束后,取出反应产物,用磁铁分离并用无水乙醇和蒸馏水交替洗涤3次,用磁铁将产物分离后放在真空干燥箱中60 ℃干燥12 h备用。
1.2.5 Fe3O4/Au纳米颗粒的合成
不添加APTES,采用浸渍法合成Fe3O4/Au。取2 g Fe3O4悬浮液分散于100 g无水乙醇中,将溶液转至250 mL三口烧瓶中。机械搅拌并超声分散,加入2 mL去离子水以及0.6 mL氨水,持续搅拌形成A液。取1 mL 0.1 mg·mL-1氯金酸的乙醇溶液分散至16 g无水乙醇中,形成B液。在机械搅拌以及超声条件下将B液缓慢滴加至A液中。滴加完毕后,关超声并持续搅拌12 h。反应结束后,分别用无水乙醇和蒸馏水交替洗涤产物3次,用磁铁将黑色沉淀物分离后再分散至60 g去离子水中。最后再将溶液转至三口烧瓶中,机械搅拌辅助超声,加入5 mL NaBH4(0.1 mol·L-1),持续反应15 min后,取出产物,用去离子水洗涤3次,用磁铁将沉淀物分离后将产物放在真空干燥箱中60 ℃干燥12 h备用。
2 结果与讨论
2.1 XRD分析
图 1为所有产物的XRD图。从图中可知,位于2θ=18.66°,30.21°,35.52°,43.27°,53.64°,57.06°和62.7°的衍射峰分别与 (111)、(220)、(311)、(400)、(422)、(511) 以及 (440) 对应,这说明样品是纯Fe3O4,且为尖晶石结构的面心立方 (FCC) 结构 (JCPDS 19-629)[20],显示出较好的结晶度。参照纳米Au的标准衍射图 (JCPDS 65-2870),并未在产物Au/Fe3O4中发现Au的特征峰,这可能是因为以下原因:(1) 金的含量太低,导致其峰强被掩盖;(2) Au主要分布在Fe3O4的表面,且其颗粒较小,高度分散,没有团聚成较大尺寸的Au。
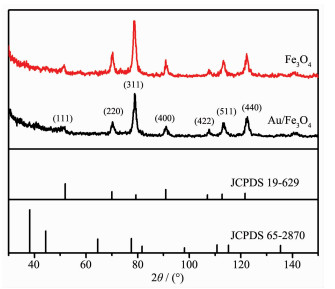 图 1
Fe3O4和Au/Fe3O4的XRD图
Figure 1.
XRD patterns of Fe3O4 and Au/Fe3O4
图 1
Fe3O4和Au/Fe3O4的XRD图
Figure 1.
XRD patterns of Fe3O4 and Au/Fe3O4
2.2 形貌分析
产物的形貌通过SEM和HRTEM确定。图 2(a)和(b)为Au/Fe3O4的SEM图,可以清楚地看出产物呈球形,具有良好的单分散性,粒径均一,其尺寸约为240 nm。由于Fe3O4是由粒径十几纳米的一级小球堆积而成的二级小球,所以其表面不光滑[21]。图 2(c)~(e)为Au/Fe3O4的HRTEM图。图 2(c)表明样品确实是单分散的球形颗粒,直径约为240 nm,这与SEM的结果一致。典型的Fe3O4微球的HRTEM图像如图 2(d)和(e),即Fe3O4微球具有2种结构:结晶度较好的中间区域和无定形结构的边缘。在无定形的边缘,可以清楚的看到它是由许多一级磁铁矿纳米小球组成的。图 2(e)中存在清晰的平行晶格条纹,这表明Fe3O4纳米晶体具有较高的结晶度。而在插图中观察到的晶格条纹为约0.48 nm,这与Fe3O4纳米晶体的 (111) 晶面一致[22]。Au/Fe3O4的粒径分析如图 2(f)所示,该结果表明Au/Fe3O4的粒径主要集中在250 nm左右,该结果与SEM和HRTEM结果一致。
 图 2
Fe3O4 (a) 和Au/Fe3O4 (b) 的SEM图像, Fe3O4@APTES-Au的HRTEH图像 (c~e) 和Au/Fe3O4的尺寸分布图 (f)
Figure 2.
SEM image of Fe3O4 (a) and Au/Fe3O4 (b), the HRTEH image of Au/Fe3O4 (c~e) and the corresponding size distribution of the Au/Fe3O4 (f)
图 2
Fe3O4 (a) 和Au/Fe3O4 (b) 的SEM图像, Fe3O4@APTES-Au的HRTEH图像 (c~e) 和Au/Fe3O4的尺寸分布图 (f)
Figure 2.
SEM image of Fe3O4 (a) and Au/Fe3O4 (b), the HRTEH image of Au/Fe3O4 (c~e) and the corresponding size distribution of the Au/Fe3O4 (f)
2.3 能谱分析
图 3为Au/Fe3O4纳米颗粒的EDS Mapping图。其中,图 3(a)显示出样品在测定区域范围内的SEM图像;在图 3(b)~(f)中,不同颜色的图像分别表示样品中的元素Au、N、C、Fe和O的富集区,所有元素均呈现出良好的分布。其中,N和C来自APTES中的甲氧基以及氨基,这就证明了APTES已经成功包覆在Fe3O4的表面;Au的mapping图表明Au已经成功通过APETS作为中间桥键成功地负载在Fe3O4的表面上。
 图 3
Au/Fe3O4的EDS Mapping图
Figure 3.
Image of Elemental mapping spectra of Au/Fe3O4
图 3
Au/Fe3O4的EDS Mapping图
Figure 3.
Image of Elemental mapping spectra of Au/Fe3O4
2.4 XPS分析
Au/Fe3O4的XPS图如图 4所示。图 4(a)为Au/Fe3O4的XPS全谱,可以看出在样品表面含有的主要元素有:Fe、O、C、N、Si以及Au,通过各元素的物质的量分数可计算出样品表面Au的质量分数为1.24%,而通过ICP-AES测试发现,在整个样品中Au的质量分数为0.45%。对比两者的结果,元素Au在表相的含量远远大于其在体相的含量,这表明大多数Au分布在Fe3O4表面。图 4(b)表明Au4f7/2和Au4f5/2这2个特征峰的结合能分别为83.80和88.30 eV,Au4f峰的出现表明金为零价[9]。图 4(c)中,Fe2p在725.1和710.6 eV出现的表观双峰归因于Fe2p1/2和Fe2p3/2,这接近Fe3O4的标准数据[23]。图 4(d)可看出Fe3O4@APTES与Au/Fe3O4中的N1s峰分别出现在398.85和399.6 eV,其中前者属于包裹在Fe3O4表面上APTES中的氨基的N原子,而后者则来自于与Au NPs配位后的N原子[24]。XPS的结果与SEM、XRD以及EDS Mapping的结果一致。
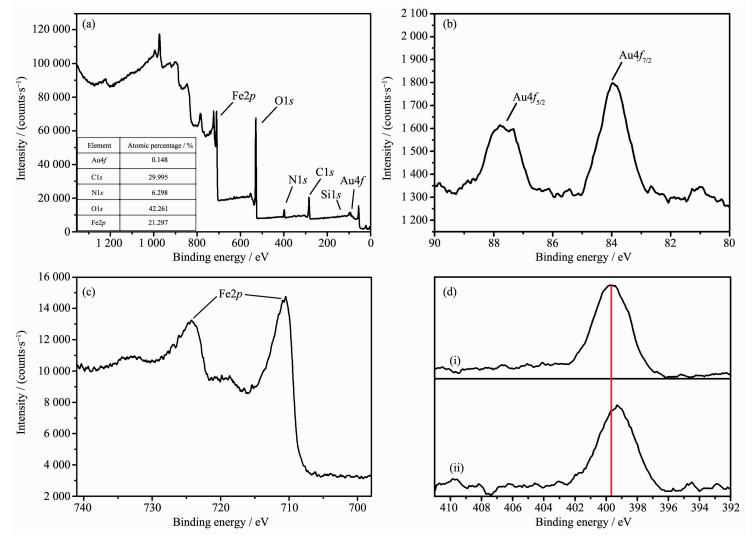 图 4
Au/Fe3O4的XPS图 (a), Au/Fe3O4中Au4f (b), Fe2p (c) 和分别来自Fe3O4@APTES (ⅰ) 与Au/Fe3O4 (ⅱ) 中N1s (d) 的高分辨谱图
Figure 4.
XPS survey spectra of Au/Fe3O4 (a) and High-resolution core-level spectra of Au4f (b), Fe2p (c) fitting of Au/Fe3O4 and N1s (d) fitting of Fe3O4@APTES (ⅰ) and Au/Fe3O4 (ⅱ)
图 4
Au/Fe3O4的XPS图 (a), Au/Fe3O4中Au4f (b), Fe2p (c) 和分别来自Fe3O4@APTES (ⅰ) 与Au/Fe3O4 (ⅱ) 中N1s (d) 的高分辨谱图
Figure 4.
XPS survey spectra of Au/Fe3O4 (a) and High-resolution core-level spectra of Au4f (b), Fe2p (c) fitting of Au/Fe3O4 and N1s (d) fitting of Fe3O4@APTES (ⅰ) and Au/Fe3O4 (ⅱ)
2.5 BET分析
图 5分别为Fe3O4和Au/Fe3O4的氮气吸附-脱附曲线图。从BET结果可知Fe3O4和Au/Fe3O4的比表面积和总体孔容积分别为83、89 m2·g-1和0.07、0.15 cm3·g-1。负载前后,Fe3O4的比表面积并未发生太大的变化,而孔容积却大大提高了。对比脱附等温线BJH (Barret-Joyner-Halenda) 的孔径分布图,可以看出Fe3O4只有一种孔径,主要分布在3.6 nm左右;而在Au/Fe3O4中,既存在介孔 (3.6 nm) 又存在大孔 (20~90 nm)。
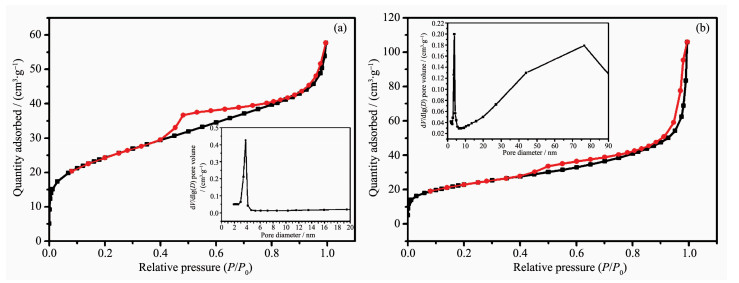 图 5
Fe3O4 (a) 和Au/Fe3O4 (b) 的N2吸附-脱附
Figure 5.
Nitrogen adsorption-desorption of Fe3O4 (a) and Au/Fe3O4 (b)
图 5
Fe3O4 (a) 和Au/Fe3O4 (b) 的N2吸附-脱附
Figure 5.
Nitrogen adsorption-desorption of Fe3O4 (a) and Au/Fe3O4 (b)
2.6 磁性分析
通过振动探针式磁强计测得所制备样品的磁滞回线,如图 6所示,Fe3O4的饱和磁化强度高达为67.35 emu·g-1,具有良好的顺磁性。而包裹了一层APTES的Fe3O4的饱和磁化强度为39.25 emu·g-1。总体而言,两个样品都具有良好的顺磁性,将制备的样品分散在无水乙醇中形成悬浮液后,通过外加磁场的作用下可快速分离收集样品,使其具有较好的可重复利用性。
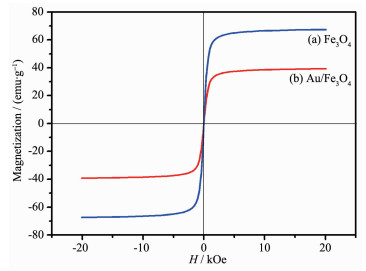 图 6
Fe3O4和Au/Fe3O4的磁滞回线图
Figure 6.
Room-temperature magnetization curves of Fe3O4 and Au/Fe3O4
图 6
Fe3O4和Au/Fe3O4的磁滞回线图
Figure 6.
Room-temperature magnetization curves of Fe3O4 and Au/Fe3O4
2.7 催化性能
由于供体对和受体对的氧化还原电位存在巨大差异,即EH3BO3(aq.)/BH4(aq.)=-1.33 V和E4-NP (aq.)/4-AP (aq.)=0.76 V,通过NaBH4将4-NPs还原成4-APs在动力学上是无法自发进行的[25]。在催化剂存在的条件下,可以克服从给体BH4-到受体4-NPs的电子转移的动力学限制,加快反应速率[26]。如图 7(a)所示,4-NPs的水溶液在317 nm处显示吸收峰。加入NaBH4后,4-NPs中的-OH基团被去质子化,在碱性条件下形成4-硝基苯酚盐阴离子,吸收峰从317 nm移到400 nm[27]。此时,溶液的颜色从淡黄色变为亮黄色。图 7(b)为Au/Fe3O4催化还原4-NPs的结果,从图中可知,当加入Au/Fe3O4后,仅需要8 min,反应就全部进行完毕,表明其具有较高的催化活性。图 7(c)~(f)为Au/Fe3O4-1、Fe3O4-Au、Fe3O4/Au和Fe3O4作为催化剂催化还原4-NPs的结果。如图所示,纯Fe3O4的催化活性很差;通过分步法制得的Au/Fe3O4-1需要14 min才能使反应进行完毕,而Fe3O4-Au和Fe3O4/Au催化反应完成的时间分别为53和140 min。这是因为分步法得到的Au/Fe3O4-1中Au的单载量较低;使用掺杂法得到的Fe3O4-Au,Au在载体表面分布不均匀,在反应过程中无法保证活性中心与反应物的充分接触;不使用APTES作为中间桥键,Fe3O4/Au中的Au无法稳定的固定在Fe3O4表面,使其很容易在洗涤中脱落或者直接团聚成较大的颗粒造成其催化活性降低。
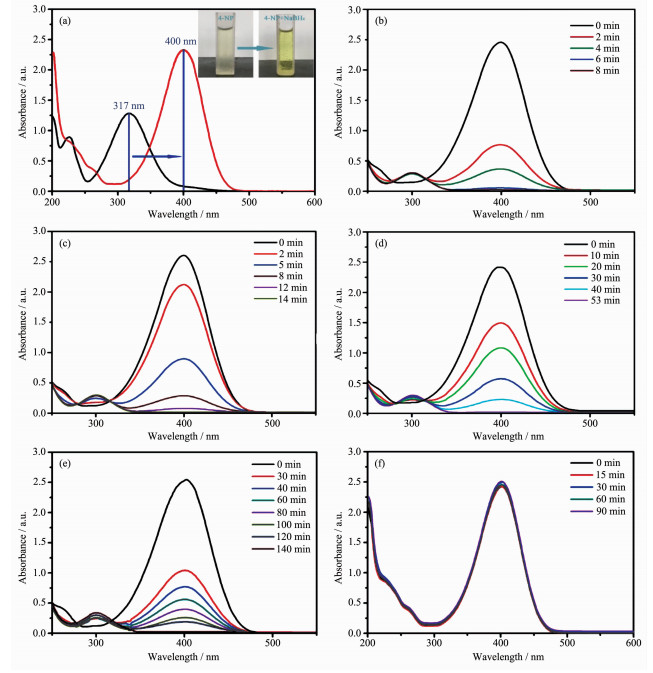 图 7
加入NaBH4前后4-NP的紫外-可见光光谱 (a) 以及Au/Fe3O4 (b), Au/Fe3O4-1 (c), Fe3O4/Au (d), Fe3O4-Au (e), Fe3O4 (f) 作为催化剂催化还原4-NP的紫外-可见光光谱
Figure 7.
UV-Vis spectra of 4-NP before and after the addition of catalyst (a) and UV-Vis absorption spectra of the reduction of 4-NP by using Au/Fe3O4 (b), Au/Fe3O4-1 (c), Fe3O4/Au (d), Fe3O4-Au (e), Fe3O4 (f) as catalyst
图 7
加入NaBH4前后4-NP的紫外-可见光光谱 (a) 以及Au/Fe3O4 (b), Au/Fe3O4-1 (c), Fe3O4/Au (d), Fe3O4-Au (e), Fe3O4 (f) 作为催化剂催化还原4-NP的紫外-可见光光谱
Figure 7.
UV-Vis spectra of 4-NP before and after the addition of catalyst (a) and UV-Vis absorption spectra of the reduction of 4-NP by using Au/Fe3O4 (b), Au/Fe3O4-1 (c), Fe3O4/Au (d), Fe3O4-Au (e), Fe3O4 (f) as catalyst
NaBH4的浓度远远大于4-NPs的浓度,所以该反应可被认为是一级动力学反应,即4-硝基苯酚盐离子的吸光度与其反应速率常数成正比,Au/Fe3O4催化反应的速率常数使用下式确定:-ln (At /A0)=kt,其中At和A0分别是在反应时间t和0 min下溶液的吸光度 (λ=400 nm)[28]。当催化剂存在时,反应的速率常数 (k) 由-ln (At /A0) 相对于时间的相应线性图的斜率确定。而k值的大小可直接反映反应的速率,即k值越大,反应速率越大,催化剂的活性也就越高[29]。如图 8和表 1,表明Au/Fe3O4的k值远远大于其他几种催化剂,这也就证明其催化活性远远大于其他的催化剂,其原因主要是以下几点:(1) 使用APTES作为有机桥键,利用氨基与氯金酸根较强的静电吸引离将Au紧紧固定在Fe3O4表面,使其不容易脱落以及团聚。(2) Fe3O4的比表面积较大,为活性中心和反应物提供了更多的接触位点,使反应更快的进行。
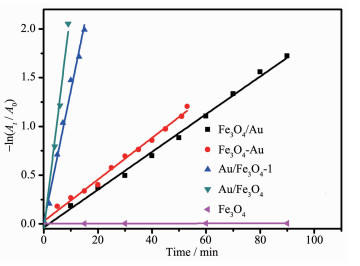 图 8
不同催化剂催化还原4-NPs反应的-ln (At /A0) 与时间的关系
Figure 8.
Plots of-ln (At /A0) versus time for the reduction of 4-NP by NaBH4 in the presence of different catalysts
图 8
不同催化剂催化还原4-NPs反应的-ln (At /A0) 与时间的关系
Figure 8.
Plots of-ln (At /A0) versus time for the reduction of 4-NP by NaBH4 in the presence of different catalysts
 表 1
不同催化剂催化反应的反应时间以及对应的反应数率常数
Table 1.
Reaction time and rate constants for the model reaction catalyzed by different catalysts
表 1
不同催化剂催化反应的反应时间以及对应的反应数率常数
Table 1.
Reaction time and rate constants for the model reaction catalyzed by different catalysts
Catalyst Reaction time/min k/min-1 Fe3O4 - 0 Fe3O4/Au 53 0.021 4 Fe3O4—Au 140 0.018 4 Au/Fe3O4-1 15 0.135 2 Au/Fe3O4 8 0.225 6 表 1 不同催化剂催化反应的反应时间以及对应的反应数率常数
Table 1. Reaction time and rate constants for the model reaction catalyzed by different catalystsAu/Fe3O4的重复性测试结果如图 9所示。重复使用反应9次后,4-NPs的转化率仍可达94%,表明Au/Fe3O4纳米颗粒显示出良好的稳定性和重复性。
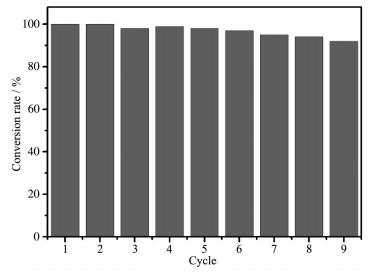 图 9
Au/Fe3O4催化还原4-NP的循环次数以及对应的转化率
Figure 9.
Conversion rates of 4-nitrophenol to 4-aminophenol for the catalytic reaction by Au/Fe3O4 nanoparticles as catalyst
图 9
Au/Fe3O4催化还原4-NP的循环次数以及对应的转化率
Figure 9.
Conversion rates of 4-nitrophenol to 4-aminophenol for the catalytic reaction by Au/Fe3O4 nanoparticles as catalyst
3 结论
本文采用简易的超声法以及原位还原法成功合成了具有高催化活性,稳定性以及可重复使用的Au/Fe3O4纳米颗粒。通过APTES作为有机桥键将Au0固定在Fe3O4表面而不团聚。Au/Fe3O4纳米颗粒显示出优异的还原4-NPs催化活性和稳定性,其反应遵循第一动力学反应,且反应速率常数为0.225 6 min-1。该催化剂拥有较高的饱和磁化强度,可在外界磁场存在的情况下快速分离,因此便于催化剂的重复利用。而通过重复性测试,可知其反应9个循环后,转化率仍高达94%。总之,Au/Fe3O4纳米颗粒易于合成,具有良好的催化活性且可回收,显示出较大的应用价值。
-
-
[1]
Haruta M, Yamada N, Iijima S, et al. J. Catal., 1989, 115:301-309 doi: 10.1016/0021-9517(89)90034-1
-
[2]
Yang X F, Wang A Q, Zhang T, et al. Acc. Chem. Res., 2013, 46:1740-1748 doi: 10.1021/ar300361m
-
[3]
解林艳, 李群艳, 王志宏, 等.无机化学学报, 2010, 26(10):1756-1760 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20101006&journal_id=wjhxxbcnXIE Lin-Yan, LI Qun-Yan, WANG Zhi-Hong, et al. Chinese J. Inorg. Chem., 2010, 26(10):1756-1760 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20101006&journal_id=wjhxxbcn
-
[4]
黄琪惠, 张豹山, 姬广斌, 等.无机化学学报, 2012, 28(10):2077-2082 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20121008&journal_id=wjhxxbcnHUANG Qi-Hui, ZHANG Bao-Shan, JI Guang-Bin, et al. Chinese J. Inorg. Chem., 2012, 28(10):2077-2082 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20121008&journal_id=wjhxxbcn
-
[5]
刘冰, 王德平, 姚爱华, 等.无机化学学报, 2008, 23(1):33-38 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20140211&journal_id=wjhxxbcnLIU Bing, WANG De-Ping, YAO Ai-Hua, et al. Chinese J. Inorg. Chem., 2008, 23(1):33-38 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20140211&journal_id=wjhxxbcn
-
[6]
Wu W, He Q G, Nie L B, et al. Nanotechnology, 2007, 18:145609-145617 doi: 10.1088/0957-4484/18/14/145609
-
[7]
刘勇健, 陈勇兵, 刘敏, 等.无机化学学报, 2015, 31(6):1165-1170 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150615&journal_id=wjhxxbcnLIU Yong-Jian, CHEN Yong-Bing, LIU Min, et al. Chinese J. Inorg. Chem., 2015, 31(6):1165-1170 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20150615&journal_id=wjhxxbcn
-
[8]
肖旺钏, 王叶敏, 王仁章, 等.无机化学学报, 2014, 30(11):2259-2563 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20141115&journal_id=wjhxxbcnXIAO Wang-Chuan, WANG Ye-Min, WANG Ren-Zhang, et al. Chinese J. Inorg. Chem., 2014, 30(11):2259-2563 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20141115&journal_id=wjhxxbcn
-
[9]
Cui L R, Lin H M, Qu F Y, et al. Eur. J. Inorg. Chem., 2014, 36:6156-6164
-
[10]
Rostamizadeh S, Estiri H, Azad M. J. Iranian Chem. Soc., 2016, 18:1367-1374
-
[11]
Zhou Y, Wang B W, Zhu X Q, et al. Nanotechnology, 2016, 27:215301-215310 doi: 10.1088/0957-4484/27/21/215301
-
[12]
Liu W J, Jiang H, Yu H Q, et al. Green Chem., 2014, 16:4198-4205 doi: 10.1039/C4GC00599F
-
[13]
Wang Z Z, Zhai S R, An Q D, et al. Eur. J. Inorg. Chem., 2015, 10:1692-1699
-
[14]
Wang M, Tian D, Yuan L J, et al. Appl. Surf. Sci., 2013, 283:389-395 doi: 10.1016/j.apsusc.2013.06.120
-
[15]
Fernandez J, Bandara J, Kiwi A J, et al. Langmuir, 1999, 15:185-192 doi: 10.1021/la980382a
-
[16]
Pany S, Parida K M, Naik B. RSC Adv., 2013, 3:4976-4984 doi: 10.1039/c3ra22775h
-
[17]
Xiong Z G, Zhang L L, Zhao X S, et al. Chem. Commun., 2010, 46:6099-6101 doi: 10.1039/c0cc01259a
-
[18]
Zheng J M, Deng Y L, Chen X G, et al. Nanoscale, 2013, 5:4894-4901 doi: 10.1039/c3nr01075a
-
[19]
Gazi S, Ananthakrishnan R. Appl. Catal. B, 2011, 105:317-325 doi: 10.1016/j.apcatb.2011.04.025
-
[20]
Deng H, Li X L, Li Y D, et al. Angew. Chem., 2005, 117:2842-2845 doi: 10.1002/(ISSN)1521-3757
-
[21]
Liu J, Sun Z K, Zhao D Y, et al. Angew. Chem. Int. Ed., 2009, 48:5875-5879 doi: 10.1002/anie.v48:32
-
[22]
Dong F P, Guo W P, Ha C S. J. Nanopart. Res., 2012, 14:1303-1311 doi: 10.1007/s11051-012-1303-9
-
[23]
Black D, Gao Y, Yang H, et al. Nano Lett., 2003, 3:261-264 doi: 10.1021/nl025918y
-
[24]
Li D X, He Q, Li J B, et al. Chem. Mater., 2007, 19:412-417 doi: 10.1021/cm062290+
-
[25]
Saha S, Pal A, Pal T, et al. Langmuir, 2010, 26:2885-2893 doi: 10.1021/la902950x
-
[26]
Gangula A, Podila R, Rao A M, et al. Langmuir, 2011, 27:15268-15274 doi: 10.1021/la2034559
-
[27]
Konar S, Kalita H, Pathak A, et al. J. Catal., 2016, 336:11-22 doi: 10.1016/j.jcat.2015.12.017
-
[28]
Perez-Lorenzo M, Liz-Marzan L M, Ballauff M, et al. Chem. Soc. Rev., 2012, 41:5577-558 doi: 10.1039/c2cs35029g
-
[29]
Du C, He S J, Chen W, et al. ChemCatChem, 2016, 8:1-6 doi: 10.1002/cctc.v8.1
-
[1]
-

图 2 Fe3O4 (a) 和Au/Fe3O4 (b) 的SEM图像, Fe3O4@APTES-Au的HRTEH图像 (c~e) 和Au/Fe3O4的尺寸分布图 (f)
Figure 2 SEM image of Fe3O4 (a) and Au/Fe3O4 (b), the HRTEH image of Au/Fe3O4 (c~e) and the corresponding size distribution of the Au/Fe3O4 (f)

图 4 Au/Fe3O4的XPS图 (a), Au/Fe3O4中Au4f (b), Fe2p (c) 和分别来自Fe3O4@APTES (ⅰ) 与Au/Fe3O4 (ⅱ) 中N1s (d) 的高分辨谱图
Figure 4 XPS survey spectra of Au/Fe3O4 (a) and High-resolution core-level spectra of Au4f (b), Fe2p (c) fitting of Au/Fe3O4 and N1s (d) fitting of Fe3O4@APTES (ⅰ) and Au/Fe3O4 (ⅱ)

图 5 Fe3O4 (a) 和Au/Fe3O4 (b) 的N2吸附-脱附
Figure 5 Nitrogen adsorption-desorption of Fe3O4 (a) and Au/Fe3O4 (b)

图 6 Fe3O4和Au/Fe3O4的磁滞回线图
Figure 6 Room-temperature magnetization curves of Fe3O4 and Au/Fe3O4

图 7 加入NaBH4前后4-NP的紫外-可见光光谱 (a) 以及Au/Fe3O4 (b), Au/Fe3O4-1 (c), Fe3O4/Au (d), Fe3O4-Au (e), Fe3O4 (f) 作为催化剂催化还原4-NP的紫外-可见光光谱
Figure 7 UV-Vis spectra of 4-NP before and after the addition of catalyst (a) and UV-Vis absorption spectra of the reduction of 4-NP by using Au/Fe3O4 (b), Au/Fe3O4-1 (c), Fe3O4/Au (d), Fe3O4-Au (e), Fe3O4 (f) as catalyst

图 8 不同催化剂催化还原4-NPs反应的-ln (At /A0) 与时间的关系
Figure 8 Plots of-ln (At /A0) versus time for the reduction of 4-NP by NaBH4 in the presence of different catalysts

图 9 Au/Fe3O4催化还原4-NP的循环次数以及对应的转化率
Figure 9 Conversion rates of 4-nitrophenol to 4-aminophenol for the catalytic reaction by Au/Fe3O4 nanoparticles as catalyst
表 1 不同催化剂催化反应的反应时间以及对应的反应数率常数
Table 1. Reaction time and rate constants for the model reaction catalyzed by different catalysts
Catalyst Reaction time/min k/min-1 Fe3O4 - 0 Fe3O4/Au 53 0.021 4 Fe3O4—Au 140 0.018 4 Au/Fe3O4-1 15 0.135 2 Au/Fe3O4 8 0.225 6  下载: 导出CSV
下载: 导出CSV
-











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 1110
- HTML全文浏览量: 178
 下载:
下载:
