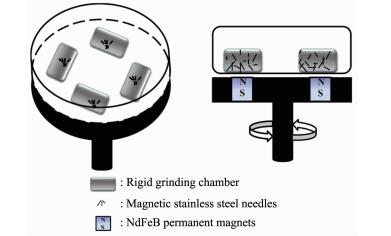
 图 1
磁力研磨机装置图
Figure 1.
Schematic diagram of the magnetic grinding method
图 1
磁力研磨机装置图
Figure 1.
Schematic diagram of the magnetic grinding method
引用本文:
季海滨, 宋沙沙, 杨玉燕, 刘洋, 赵增典. 低氧掺杂纳米石墨片的制备及其电化学性能[J]. 无机化学学报,
2016, 32(5): 745-752.
doi:
10.11862/CJIC.2016.107

Citation: JI Hai-Bin, SONG Sha-Sha, YANG Yu-Yan, LIU Yang, ZHAO Zeng-Dian. Low Oxygen-Doped Graphite Nanosheet: Preparation and Electrochemical Performance[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(5): 745-752. doi: 10.11862/CJIC.2016.107
Citation: JI Hai-Bin, SONG Sha-Sha, YANG Yu-Yan, LIU Yang, ZHAO Zeng-Dian. Low Oxygen-Doped Graphite Nanosheet: Preparation and Electrochemical Performance[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(5): 745-752. doi: 10.11862/CJIC.2016.107
低氧掺杂纳米石墨片的制备及其电化学性能
English
Low Oxygen-Doped Graphite Nanosheet: Preparation and Electrochemical Performance
Abstract:
A new physics method of magnetic grinding was provided to produce nanographite, which can efficiently break flake graphite to nanoscale. The obtained graphite nanosheets possessed oxygen-containing groups and it's specific surface area could reach to 804 m2·g-1 with increase of grinding time. The electrochemical data showed that the capacitance of the graphite nanosheet was 266.8 F·g-1 under the current density of 0.1 A·g-1, indicating excellent electrochemical performance.
-
Key words:
- magnetic grinding
- / graphite nanosheet
- / capacitance
-
自石墨烯被发现以来,由于其具有优良的导热、导电、高强度、透光性、高比表面积[1-3]等性能,引起了科学家的研究热潮。例如,在超级电容器的研究中,一般将超级电容器分为双电层和赝电容[4],由于石墨烯具有极好的电子传导能力[5],其理论比表面积可达2 630 m2·g-1[6],可以应用于超级电容器中。石墨烯超级电容器具有充放电速率快,能量密度高等的特点,其理论比电容量为329 F·g-1 [7-8]。但是,石墨烯的团聚现象[9],大大限制了其电化学性能。
石墨烯研究至今,科学家们一直在探索一种高效、低成本的制备石墨烯的方法。最初,英国曼彻斯特大学安德烈-海姆教授用胶带将石墨层层剥离制备了稳定存在的石墨烯[10]。在随后的探索中,科学家们探究了多种剥离石墨制备石墨烯的方法,常见的化学法如Hummers法[11]、有机合成法[12]、电弧放电法[13]、溶剂热法[14]、化学气相沉积法[15]、改良Hummers法[16]等一系列化学方法,将石墨烯的认识和探究提升到一个新的领域。化学法也是至今制备石墨烯最为普遍的一种方法[17-18],但化学法制备石墨烯的过程中对工艺要求较高或需要大量的化学试剂,限制了其工业生产应用。相对于化学法,物理法剥离石墨没有复杂的化学试剂 (如:硫酸、硝酸,高锰酸钾等) 的参与,对环境零污染,更贴近于工业生产,球磨法[19]是一种较为常规的制备纳米石墨片 (graphite nanoseets,GNS) 的物理方法,经球磨制备得到的纳米石墨材料的厚度能降低至数十层。本文采用了一种磁力研磨的方法制备纳米石墨片,该方法是由高速旋转的磁场带动弱磁性的钢针,在固定的研磨腔内通过钢针间相互的碰撞与摩擦,对石墨进行有效的切割、剥离。相比传统的球磨法,磁力研磨法效率更高,方法简单,可以实现高效批量的生产纳米石墨片。经磁力研磨后的纳米石墨片厚度小,具有较高的比表面积。因此,本实验用磁力研磨机在空气气氛下对石墨进行剥离,制备了得到了低氧含量掺杂的纳米石墨片,利用扫描电子显微镜 (SEM)、透射电子显微镜 (TEM)、N2吸-脱附实验、X射线衍射 (XRD) 表征了纳米石墨片的形貌和孔结构;通过红外光谱测试 (FT-IR)、热重分析 (TGA)、X射线电子能谱分析 (XPS)、元素分析等对制备得到的纳米石墨片的表面官能团进行了表征,并探索了其电化学性能。
1 实验部分
1.1 磁力研磨法制备石墨烯纳米片
以天然鳞片石墨为原料,用电子天平称取一定质量的弱磁性钢针置于研磨腔体内密封,石墨与钢针质量比为50:1,设置磁力研磨机转速、时间,用磁力研磨机分别研磨2、4、6、8、12、16、20 h,研磨完成后,用300目筛网将石墨与磁性钢针分离。将筛分的石墨烯纳米片倒入1 mol·L-1稀盐酸中,磁力搅拌24 h,用蒸馏水反复洗涤至中性,然后,在100℃干燥12 h,根据研磨时间,将样品分别记为GNS 2、GNS 4、GNS 6、GNS 8、GNS 12、GNS 16和GNS 20。
1.2 材料表征及电化学性能测试
1.2.2 电极制备与电化学测试
称取一定质量的石墨电极材料,以5%(w/w) 的聚四氟乙烯乳液为粘结剂,均匀地涂在钛网上,在300 kg·cm-2压力下制成工作电极,在100 ℃干燥后称量并计算电极材料的质量,将工作电极在1 mol·L-1的H2SO4电解液中抽真空浸润30 min再进行电化学测试。所有电化学测试采用CHI760E电化学工作站进行测试,循环伏安曲线和恒电流充放电,以及交流阻抗均采用三电极测试体系,以铂片电极为对电极,饱和甘汞电极作为参比电极。
1.2.1 材料表征
材料的微观形貌采用Sirion 200 FEI (SEM,Holand) 扫描电子显微镜观察分析,分辨率1.5 nm,加速电压为0.2~30 kV;透射电子显微镜 (TEM,Tecnai G2 F20 S-TWIN FEI,USA) 点分辨率小于0.24,加速电压20~200 kV;氮气吸附等温线通过美国ASAP 2020M氮气吸附仪测定,用BET法计算得到材料的比表面积;材料晶体结构使用X射线衍射 (XRD,D8Advance,Germany) 进行表征,Cu靶Kα射线,波长0.150 4 nm,测试电压40 kV,电流35 mA,扫描范围3°~60°,步长是0.02°;用傅里叶转换红外光谱 (FT-IR,Nicolet 5700,USA)、X射线光电子能谱分析 (XPS,PHI 5000 Versa Probe,Japan)、热失重测试仪 (TG,DSCQ100,USA) 表征材料的表面性质。
2 结果与讨论
如图 1所示,磁力研磨机是在电机轴驱动下,带动强磁铁不断地旋转,在一定区域内产生不断变换的磁场,以此带动弱磁性针状研磨介质在固定的研磨腔内相互剧烈碰撞、摩擦,在这过程中产生的剪切力和摩擦力会破坏石墨层与层之间的范德华力,石墨C=C化学键力,对石墨进行有效的破碎、剥离。随着研磨时间的增加,石墨的尺寸和厚度都会不断的减小,当被剥离的石墨尺寸达到纳米级的时候,这些具有较高活性的纳米石墨片又重新团聚在一起,形成相对较大的纳米石墨团簇体,之后团簇体被机械力打开又重新团聚。传统球磨法剥离纳米石墨,是由研磨腔的高速运动来带动研磨介质运动,往往通过铁质、玛瑙或其他材质的球形研磨介质之间相互碰撞挤压来破碎和剥离石墨,而磁力研磨法的研磨腔是固定的,其研磨介质在研磨腔内是主动运动,所以研磨介质的运行状态是不同的,并且磁力研磨的研磨介质是0.3 cm×5 cm细长圆柱形弱磁性钢针,在强磁场力带动下高速旋转和运动,当两个高速运转钢针之间直接碰撞或摩擦时,会在瞬间产生较强作用力,当多个钢针相互碰撞的时候,会产生多个作用点,这样就增加了研磨介质与石墨之间的接触概率,所以极大提高了研磨效率。
图 1
磁力研磨机装置图
Figure 1.
Schematic diagram of the magnetic grinding method
图 2为研磨不同时间的纳米石墨片 (GNS) 扫描电镜图。从图 2a中可以看出,初始磷片石墨由于范德华作用力有序紧密的堆叠在一起,具有较大的尺寸和厚度。石墨研磨6 h后 (图 2b) 已完全找不到较大尺寸的石墨,并且研磨后的纳米石墨尺寸大小均一,基本在5 μm以内,这说明研磨的效率是极高的。由于石墨尺寸的突然降低,形成了大小不一的团簇体,在团簇体边缘 (图 2c) 看到近乎透明且呈卷曲状态类似于石墨烯片的GNS;研磨12 h以后 (图 2d,e) 的纳米石墨片的尺寸已完全在纳米级别,在大的团簇体中发现较多的纳米石墨片相互堆叠插杂在一起,形成了较多的孔隙结构,并且纳米石墨片尺寸厚度整体上大大降低;当研磨至20 h时,被剥离的GNS边缘基本是无规则的状态,扫描电镜图中部分地方存在放电现象,说明其晶体结构破坏导电性变差,并且团聚现象也较为严重 (图 2f,g)。从研磨12 h的GNS透射电镜图 (图 2h,j) 中看到,经研磨的纳米石墨片还是具有一定厚度,但是其厚度已经在10 nm以内,并且SAED中电子经衍射后形成了光斑而非环状,也佐证了GNS具有较小的片层厚度,说明磁力研磨能够对纳米石墨片进行有效的剥离。
 图 2
不同研磨时间的GNS扫描电镜图 (a~g) 和GNS 12的透射电镜图 (h, j)
Figure 2.
SEM images of GNS at different grinding time (a~g) and TEM images of GNS 12 (h, j)
图 2
不同研磨时间的GNS扫描电镜图 (a~g) 和GNS 12的透射电镜图 (h, j)
Figure 2.
SEM images of GNS at different grinding time (a~g) and TEM images of GNS 12 (h, j)
从图 2中能够明显的看到,研磨时间越长,制得的GNS的粒径厚度越小。而石墨烯的理论比表面积是2 630 m2·g-1,但是从BET比表面变化图中,我们可以看到在8 h内,研磨的GNS比表面积迅速增加至800 m2·g-1,当研磨至12 h时,其比表面积最高可达804 m2·g-1(图 3a)。结合扫描电镜图可以认为,在研磨的过程中GNS的团簇效应是一直存在的,随着研磨时间的增长,尺寸不断减小的GNS更加容易团聚在一起,形成大小不一的团簇体。在研磨的过程中,这些团聚体被打开后又重新团聚直至达到一定的平衡,如图 3a所示,研磨8 h后其比表面积的变化趋于稳定。但研磨16 h后,其比表面积呈下降趋势,可能是长时间的研磨会较大程度地破坏GNS的晶型结构,形成无定形的碳材料。图 3b为研磨12 h的GNS的吸脱附曲线,该曲线为典型的间劈孔类型吸脱附曲线,并且发现其滞后回环并不很明显,说明GNS会相互插接形成大孔结构,从研磨12 h的GNS扫描电镜图中也能体现出来。
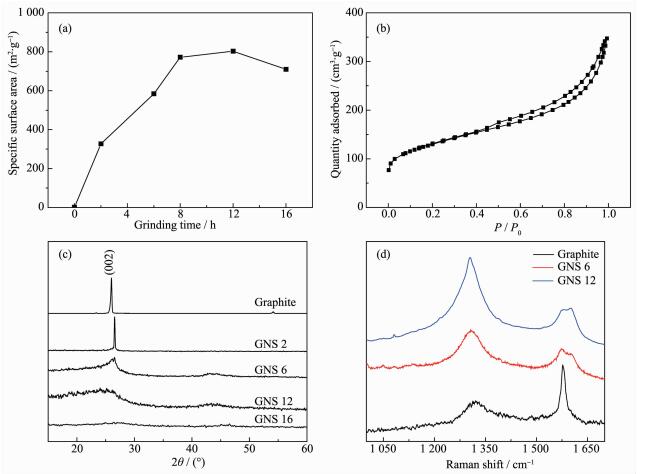 图 3
(a) 不同研磨时间GNS的BET比表面; (b) GNS 12的吸-脱附曲线; (c) 不同研磨时间GNS的XRD图
Figure 3.
(a) BET surface data of graphite ground for different time; (b) Adsorption and desorption isotherms of GNS 12; (c) Powder XRD patterns of different GNS and pristine graphite; (d) Raman spectra of pristine graphite and GNS 6 and GNS 12
图 3
(a) 不同研磨时间GNS的BET比表面; (b) GNS 12的吸-脱附曲线; (c) 不同研磨时间GNS的XRD图
Figure 3.
(a) BET surface data of graphite ground for different time; (b) Adsorption and desorption isotherms of GNS 12; (c) Powder XRD patterns of different GNS and pristine graphite; (d) Raman spectra of pristine graphite and GNS 6 and GNS 12
图 3c为石墨研磨不同时间的X射线衍射图,从图中可以看到,初始石墨在26.02°有较强的衍射峰,根据布拉格定律能够计算出相邻石墨层间距为0.34 nm。研磨2 h后,研磨的GNS在26.02°依然保持着较强的衍射峰。研磨6 h后,石墨的特征峰明显的减弱和宽化,说明研磨的GNS厚度和粒径都明显的降低。研磨12 h后发现,在26.02°的衍射峰严重宽化,可认为是被剥离的尺寸较小的GNS形成的团簇体所表现出的峰类型。当研磨时间增至20 h时,已经没有明显的石墨特征峰,说明较长时间的研磨会极大增加石墨样品的无序度,石墨的晶型结构已完全破坏[20]。图 3d是初始石墨和纳米石墨片的拉曼图谱。从图中可以看出,原始石墨和研磨时间分别为6和12 h的GNS分别在1 321、1 576 cm-1处均有明显的D峰和G峰,并且经研磨后的纳米石墨片D峰信号强度变大至超过相应的G峰信号,说明研磨介质对石墨有效的破碎和剥离致使石墨的粒径和厚度被高效减小,同时也造成了纳米石墨片的晶格缺陷,导致了D峰的强度变大。纳米石墨片的研磨时间变长,ID/IG值从石墨的0.612变化至1.217和1.632,ID/IG值的变化也体现了纳米石墨片在机械力作用下缺陷的增多和厚度粒径的减小。经磁力研磨后纳米石墨片出现了G峰的劈裂,这是应力条件下破碎剥离纳米石墨片的现象,造成该现象的原因为在机械力破碎石墨的过程中,由于机械力的作用,纳米石墨片在应力作用下C=C键长伸长产生不均匀性,导致纳米石墨片的对称性降低,对称性的降低使得在无应力下拉曼谱中G峰对应的简并的2支光学模声子劈裂开来,从而拉曼谱中G峰分裂为G+和G-两个峰。
磁力研磨是通过机械力的作用打开石墨内C=C化学键,化学键断裂之后在GNS的边缘依然会存在不稳定的电子或电子对,即悬键[21-22]。这种悬键表现出极高的活性,结合扫描电镜图 (图 2b,d),GNS纳米片的团簇可以认为是石墨纳米尺寸效应和被剥离的石墨边缘静电力共同作用的结果。GNS边缘的悬键要达到稳定的状态,一方面要与邻近的悬键相结合形成新键,另一方面,会吸引周围活泼分子中相对不稳定电子与其成键,并且与其成键的分子的外层电子越不稳定,越容易失电子与悬键形成新键。不同的气体分子与悬键结合的难易程度是不同的,所以研磨过程中,在仅有一种活泼气体气氛的条件下,GNS边缘的悬键有更大几率与该活泼分子成键,因此,我们能在制备GNS的过程中有效地增加单元素或指定元素的含量。
在特定活泼气氛条件下磁力研磨石墨,可以有效的提高特定元素在纳米石墨片中的含量,比如在空气气氛下,氮气占78%,氧气占21%,其余气体分子如水分子、二氧化碳、二氧化氮等只占微量 (包括极微量活性较高的氢气),在研磨的过程中,被破坏的GNS边缘产生的悬键会吸引研磨气氛中较活泼的气体分子 (如氧气) 或质子与之成键。从图 4a中可以清楚的看到,石墨在1 578 cm-1处有C=C双键特征吸收峰,在1 734 cm-1处有C=O的伸缩振动峰,以及1 047 cm-1和1 465 cm-1处有C-O与C-OH的伸缩振动峰[23-24];从图 4b即GNS的XPS全谱图中发现,除了C1s(~284.6 eV) 较强的吸收峰,也有明显的O1s(~531.3 eV) 峰。通过C1s高分辨XPS谱分峰拟合后发现,空气气氛下研磨的GNS存在较强的C=C (~286.4 eV)、COH (~286.5 eV) 峰和C=O (~287.9 eV)、C (O) OH (~288.9 eV) 以及较弱的C=N (~290.9 eV) 峰,在285.6 eV处的拟合峰可能为石墨破碎所造成的sp3杂化峰,由此证明了研磨后的GNS边缘存在一定量的羟基、羧基含氧官能团。图 4d是初始石墨和石墨分别研磨2、6、12 h的样品在30~100 ℃的热重分析曲线,从图中可以看出初始石墨的质量是几乎不变的。经研磨的GNS随着温度的升高,质量减少较为平缓 (其中120 ℃为样品中水分和CO2的脱除),并且研磨12 h的纳米石墨片在温度达到700 ℃时,质量残留为87%,说明石墨经研磨后虽含有一定的含氧官能团,但不能像氧化石墨达到高氧化程度。比较不同研磨时间GNS热重曲线发现,研磨时间越长,在较高温度下残余质量越低,证明长时间的研磨,能增加GNS的羟、羧基含量。因此,在控制研磨时间的前提下是能够制备低氧含量掺杂的GNS。
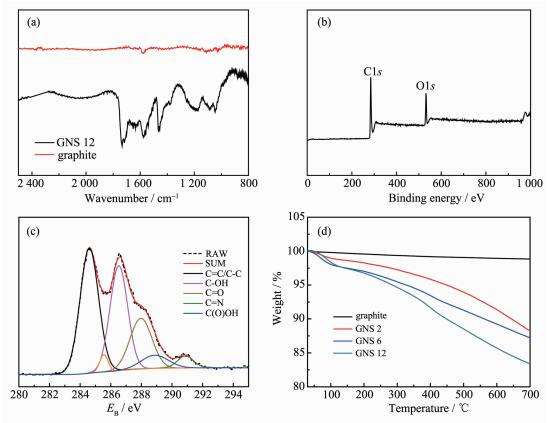 图 4
(a) 初始石墨和GNS 12的红外光谱图; (b) GNS 12的XPS全谱图; (c) GNS 12的高分辨C1s XPS谱图; (d) 初始石墨及GNS 2、GNS 6、GNS 12的热重分析曲线
Figure 4.
(a) FT-IR spectra of pristine graphite and GNS 12; (b) XPS survey spectrum of GNS 12; (c) High resolution C1s spectrum of GNS 12; (d) TGA of pristine graphite and GNS ground for GNS 2, GNS 6, GNS 12
图 4
(a) 初始石墨和GNS 12的红外光谱图; (b) GNS 12的XPS全谱图; (c) GNS 12的高分辨C1s XPS谱图; (d) 初始石墨及GNS 2、GNS 6、GNS 12的热重分析曲线
Figure 4.
(a) FT-IR spectra of pristine graphite and GNS 12; (b) XPS survey spectrum of GNS 12; (c) High resolution C1s spectrum of GNS 12; (d) TGA of pristine graphite and GNS ground for GNS 2, GNS 6, GNS 12
石墨烯具有较高的比表面积能够吸附更多的电荷,是制备双电层超级电容器的优良材料。而现在对于石墨烯超级电容器的研究往往是通过复合金属氧化物或掺杂具有氧化还原性的杂元素如氮、氧、硫等来制备复合/掺杂石墨烯超级电容器来提高电容量[25-27],由于氮、氧、硫等具有多种化学价态,在充放电的过程中,通过氧化还原反应中价态改变时电子的转移,来达到储存或释放电荷的目的。磁力研磨12 h的GNS具有较高的比表面积 (约800 m2·g-1),能够提供较好的双电层特性,并且在GNS边缘低含量的羟、羧基等含氧官能团在充放电时的氧化还原反应能够提供一定的赝电容,所以经研磨后制备的GNS具有双电层-赝电容复合超级电容器的特性。图 5a、5b分别为不同样品在扫描速率30和100 mV·s-1时的循环伏安曲线,结合BET分析,研磨2 h后样品比表面积为327 m2·g-1,石墨片层间未打开,只具有较小的电容量。研磨6 h后比表面积约600 m2·g-1,继续研磨,其比表面积保持在800 m2·g-1左右,研磨时间超过6 h后,各样品的循环伏安曲线形状基本类似,这也是由于打开的GNS团簇所导致的,这也说明磁力研磨的样品是石墨片层的剥离与GNS再团簇的共同作用的结果。各样品在扫描速率30 mV·s-1的循环伏安曲线图形总体上趋于矩形,具有良好的对称性,说明具有良好的双电层特性;同时,也能够发现由GNS边缘结合含氧官能团提供的明显的氧化还原峰,并具有较高的可逆性,这表明是以双电层和赝电容方式储存电荷。研磨12 h的GNS的循环伏安曲线 (图 5c) 在5~50 mV·s-1低扫描速率下图形接近于矩形,但同时表现出了对称的氧化还原峰,但在100 mV·s-1扫描速率下图形变形较大。对比各样品扫描速率100 mV·s-1的循环伏安曲线,都在一定程度上偏离了矩形,但其变形程度差别不大。
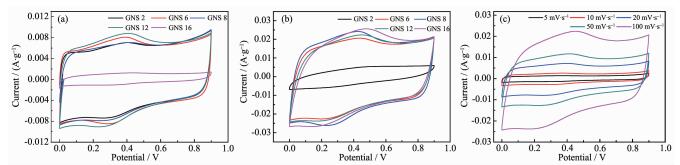 图 5
不同研磨时间GNS在30 mV·s-1 (a) 和100 mV·s-1 (b) 扫描速率下的循环伏安曲线; (c) GNS 12在不同扫描速率下的循环伏安曲线
Figure 5.
CV curves of GNS ground for different time at scan rate of 30 mV·s-1 (a) and 100 mV·s-1 (b); (c) CV curves of GNS 12 at different scan rate
图 5
不同研磨时间GNS在30 mV·s-1 (a) 和100 mV·s-1 (b) 扫描速率下的循环伏安曲线; (c) GNS 12在不同扫描速率下的循环伏安曲线
Figure 5.
CV curves of GNS ground for different time at scan rate of 30 mV·s-1 (a) and 100 mV·s-1 (b); (c) CV curves of GNS 12 at different scan rate
不同的研磨时间,对石墨的剥离以及剥离GNS边缘的含氧官能团都有不同程度的影响。图 6a为各样品在0.3 A·g-1恒电流时的充放电曲线,由图可知各样品 (除研磨2 h的石墨) 充放电曲线在一定程度上偏离等腰三角形,说明其储电机理有2种:0~0.4 V电位窗口范围内曲线为直线,应为双电层电容储电;0.4~1 V电位窗口曲线较弯,并持续时间较长,应为赝电容储能。研磨6 h以后,可以看到明显的电压降,与循环伏安曲线相吻合,说明GNS边缘的含氧官能团与电解液离子发生了法拉第反应。从图中我们还能发现,石墨剥离研磨6~12 h,样品的充放电时间是不断增加的。电容量的提高主要取决于GNS的有效比表面积所提供的双电层储能,以及破碎的GNS周围结合的含氧官能团提供的赝电容,说明在磁力研磨的过程中虽然会形成团簇体,但在保证石墨晶格结构前提下,其有效的比表面积是增加的,并且随着更多C=C键的断裂,在GNS边缘会结合更多的含氧官能团,也会增加其法拉第赝电容特性。
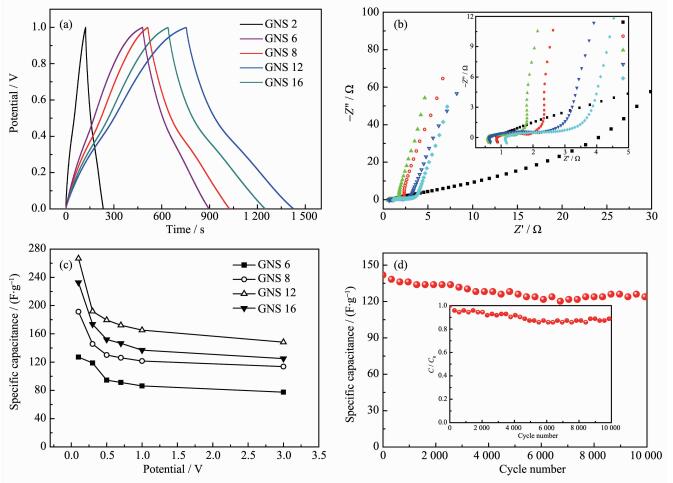 图 6
不同研磨时间石墨的恒电流充放电曲线 (a) 和交流阻抗曲线 (b); 各样品不同电流密度下比电容变化曲线 (c) 以及GNS 12在电流密度2 A·g-1下的循环寿命 (d)
Figure 6.
Galvanostatic charge/discharge curves (a) and electrochemical impedance spectra (b) of GNS ground for different time; Specific capacitance of different GNS at various current densities (c) and galvanostatic cycling performance of GNS 12 at 2 A·g-1(d)
图 6
不同研磨时间石墨的恒电流充放电曲线 (a) 和交流阻抗曲线 (b); 各样品不同电流密度下比电容变化曲线 (c) 以及GNS 12在电流密度2 A·g-1下的循环寿命 (d)
Figure 6.
Galvanostatic charge/discharge curves (a) and electrochemical impedance spectra (b) of GNS ground for different time; Specific capacitance of different GNS at various current densities (c) and galvanostatic cycling performance of GNS 12 at 2 A·g-1(d)
在三电极体系下对不同研磨时间GNS进行了交流阻抗测试,测试的频率范围是0.1~1×105 Hz,图 6b给出了不同样品的Nyquist图。研磨2 h的GNS由于剥离时间较短,依然保持着较大尺寸石墨的特性,并没有表现出双电层电容特性,曲线实部阻抗与Z'轴的截距表示等效串联电阻,主要是电解液和集流体以及电极材料之间接触的接触电阻。随着研磨时间的增加,石墨被剥离成纳米石墨进一步剥离到GNS再形成团簇体,可以看到石墨研磨8和12 h时的等效电阻小于研磨6 h GNS的等效电阻,说明长时间研磨剥离出的GNS更有利于与电解液的接触。而如研磨16 h时,过长时间的研磨对GNS晶型结构过多的破坏使其导电性变差,导致不利于离子液与其接触。高频区的半圆形弧线表示电荷转移电阻,是指电解液在电极材料内部的电荷转移电阻,弧线直径越大其电阻越大,从内插图中可以发现石墨在研磨6和8 h的电荷转移电阻相差不大。但是,当研磨时间增加,石墨尺寸、片层进一步减小,破碎的GNS之间的搭接会导致电荷转移速率变慢,因此在阻抗图中表现出较大的圆弧半径。由于长时间研磨在GNS边缘含氧官能团的增加,研磨12 h的GNS在中频区Warburg特征斜线的斜率相对较大,而研磨6和8 h的GNS在低频和中频区阻抗曲线相对更垂直于Z'轴,显示其双电层-赝电容特性。
经过恒电流充放电曲线可计算出不同样品在0.1~3 A·g-1电流密度下的比电容值,如图 6c所示,随着电流密度的增加,各样品的比电容都有不同程度的衰减。研磨时间越长,其衰减速率越快,这是由于长时间的研磨剥离出的GNS会重新团聚在一起,形成不同的团簇体。在小电流放电的时候,电解质离子有足够的时间进入到团簇体的内部,而在较大的电流密度下,电解液离子只能吸附在团簇体表面,只有部分电极表面得到利用。从图中还能够看到石墨在研磨至12 h的比电容量在各电流密度下相对最高,也说明石墨得到了有效的剥离,制备的GNS提供的双电层电容和片层边缘的含氧官能团提供的赝电容都有一定程度的增加。然而当研磨时间过长,对于石墨晶型结构破坏太严重时,其比电容反而降低。图 6d为研磨12 h的GNS的循环寿命图,在2 A·g-1电流密度下循环充放电10 000次,可以发现GNS的比电容值随着循环次数的增加而减小,这是由于在充放电的过程中,GNS边缘不稳定的官能团的分解,致使其丢失了部分赝电容,GNS 12在10 000次充放电后比电容值为123 F·g-1,可达到首次充放电的87%,具有较好的循环稳定性。
3 结论
通过磁力研磨法剥离石墨,在相对较短时间内能高效的制备晶型结构良好、比表面积高 (804 m2· g-1)、低氧含量掺杂的纳米石墨片。该方法操作简单、周期短、无需加入其他化学试剂。所制备的GNS边缘少量含氧官能团的加入,在不影响导电性能的前提下,经电化学测试表明能够提供一定的赝电容,在0.1 A·g-1电流密度下电容量为266.8 F·g-1,有效增加了该材料的电容特性。该方法是一种高效制备低含氧量纳米石墨片的物理方法,并且对研磨气氛中其他活泼元素的低含量掺杂方法也具有较好的应用前景。
-
-
[1]
Stoller M D, Park S, Zhu Y W, et al. Nano Lett., 2008, 8(10):3489-3502 doi: 10.1021/nl802558y
-
[2]
Balandin A A, Ghosh S, Bao W Z, et al. Nano Lett., 2008, 8(3):902-907 doi: 10.1021/nl0731872
-
[3]
Lee C, Wei X D, Kysar J W, Hone J. Science, 2008, 321(18):385-388 http://www.scirp.org/reference/ReferencesPapers.aspx?ReferenceID=1313019
-
[4]
Bonnefoi L, Simon P, Fauvarque J F, et al. J. Power Sources, 1999, 80:149-155 doi: 10.1016/S0378-7753(99)00069-5
-
[5]
Zhang L L, Zhou R, Zhao X S. J. Mater. Chem., 2010, 20:5983-5992 doi: 10.1039/c000417k
-
[6]
Rao C N R, Sood A K, Voggu R, et al. J. Phys. Chem. Lett., 2010, 1(2):572-580 doi: 10.1021/jz9004174
-
[7]
Wang Y, Wu Y P, Huang Y, et al. J. Phys. Chem. C, 2011, 115(46):23192-23197 doi: 10.1021/jp206444e
-
[8]
Xia J, Chen F, Li J, et al. Nat. Nanotechnol., 2009, 4(8):505-509 doi: 10.1038/nnano.2009.177
-
[9]
Ning G, Fan Z, Wang G, et al. Chem. Commun., 2011, 47:5976-5978 doi: 10.1039/c1cc11159k
-
[10]
Novoselov K S, Geim A K, Morozov S V, et al. Science, 2004, 306(5696):666-669 doi: 10.1126/science.1102896
-
[11]
Hummers W, Offeman R. J. Am. Chem. Soc., 1958, 80(6):1339 doi: 10.1021/ja01539a017
-
[12]
Chang H X, Wu H K. Adv. Funct. Mater., 2012, 23(26):1984-1997
-
[13]
Choi D H, Wang Q, Azuma Y, et al. Sci. Rep., 2013, 3:1617 doi: 10.1038/srep01617
-
[14]
Tran V K, Dong S K, Yong J K, et al. Chem. Eng. J., 2013, 232:346-355 doi: 10.1016/j.cej.2013.07.123
-
[15]
Li Y F, Li M C, Gu T S, et al. Appl. Surf. Sci., 2013, 284:207-213 doi: 10.1016/j.apsusc.2013.07.081
-
[16]
Wu X X, Xu H F, Shen Y, et al. Electrochim. Acta, 2014, 138:264-269 doi: 10.1016/j.electacta.2014.06.124
-
[17]
Si Y C, Samulski E T. Nano Lett., 2008, 8(6):1679-1682 doi: 10.1021/nl080604h
-
[18]
Compton O C, Nguyen S. Small, 2010, 6:711-723 doi: 10.1002/smll.v6:6
-
[19]
Jeon I Y, Shin Y R, Sohn G J, et al. Proc. Natl. Acad. Sci.U.S.A., 2012, 109(15):5588-5593 doi: 10.1073/pnas.1116897109
-
[20]
Zhang Y, Wedderburn J, Harris R, et al. Mater. Res. Bull., 2014, 60:648-651 doi: 10.1016/j.materresbull.2014.09.024
-
[21]
Zhang Q, Diao D F, Yang L. Surf. Coat. Technol., 2013, 237:230-233 doi: 10.1016/j.surfcoat.2013.06.039
-
[22]
Zhou K, Zhou W J, Liu X J, et al. Nano Energy, 2015, 12:510-520 doi: 10.1016/j.nanoen.2015.01.017
-
[23]
Zhang T, Zhang D, Shen M. Mater. Lett., 2009, 63(23):2051-2054 doi: 10.1016/j.matlet.2009.06.050
-
[24]
Si Y, Samulski E T. Chem. Mater., 2008, 20(21):6792-6797 doi: 10.1021/cm801356a
-
[25]
Li S M, Yang S Y, Wang Y S, et al. J. Power Sources, 2015, 278:218-229 doi: 10.1016/j.jpowsour.2014.12.025
-
[26]
Johra F T, Jung W G. Appl. Surf. Sci., 2015, 357:1911-1914 doi: 10.1016/j.apsusc.2015.09.128
-
[27]
Yang T T, Li R Y, Long X H, et al. Electrochim. Acta, 2016, 187:143-152 doi: 10.1016/j.electacta.2015.11.043
-
[1]
-

图 2 不同研磨时间的GNS扫描电镜图 (a~g) 和GNS 12的透射电镜图 (h, j)
Figure 2 SEM images of GNS at different grinding time (a~g) and TEM images of GNS 12 (h, j)
a: pristine graphite; b, c: GNS 6; d, e: GNS 12; f, g: GNS 20

图 3 (a) 不同研磨时间GNS的BET比表面; (b) GNS 12的吸-脱附曲线; (c) 不同研磨时间GNS的XRD图
Figure 3 (a) BET surface data of graphite ground for different time; (b) Adsorption and desorption isotherms of GNS 12; (c) Powder XRD patterns of different GNS and pristine graphite; (d) Raman spectra of pristine graphite and GNS 6 and GNS 12

图 4 (a) 初始石墨和GNS 12的红外光谱图; (b) GNS 12的XPS全谱图; (c) GNS 12的高分辨C1s XPS谱图; (d) 初始石墨及GNS 2、GNS 6、GNS 12的热重分析曲线
Figure 4 (a) FT-IR spectra of pristine graphite and GNS 12; (b) XPS survey spectrum of GNS 12; (c) High resolution C1s spectrum of GNS 12; (d) TGA of pristine graphite and GNS ground for GNS 2, GNS 6, GNS 12

图 5 不同研磨时间GNS在30 mV·s-1 (a) 和100 mV·s-1 (b) 扫描速率下的循环伏安曲线; (c) GNS 12在不同扫描速率下的循环伏安曲线
Figure 5 CV curves of GNS ground for different time at scan rate of 30 mV·s-1 (a) and 100 mV·s-1 (b); (c) CV curves of GNS 12 at different scan rate

图 6 不同研磨时间石墨的恒电流充放电曲线 (a) 和交流阻抗曲线 (b); 各样品不同电流密度下比电容变化曲线 (c) 以及GNS 12在电流密度2 A·g-1下的循环寿命 (d)
Figure 6 Galvanostatic charge/discharge curves (a) and electrochemical impedance spectra (b) of GNS ground for different time; Specific capacitance of different GNS at various current densities (c) and galvanostatic cycling performance of GNS 12 at 2 A·g-1(d)
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 758
- HTML全文浏览量: 181
 下载:
下载:
