Table 1.
The experimental phenomenon with different initiating systems
引用本文:
杨科, 刘强, 文帅, 徐舒心, 施晨琦. 异丁烯与对氯甲基苯乙烯正离子共聚合研究[J]. 高分子学报,
2020, 51(4): 355-365.
doi:
10.11777/j.issn1000-3304.2019.19179

Citation: Ke Yang, Qiang Liu, Shuai Wen, Shu-xin Xu, Chen-qi Shi. Study of Cationic Copolymerization of Isobutylene and p-(Chloromethyl)styrene[J]. Acta Polymerica Sinica, 2020, 51(4): 355-365. doi: 10.11777/j.issn1000-3304.2019.19179
Citation: Ke Yang, Qiang Liu, Shuai Wen, Shu-xin Xu, Chen-qi Shi. Study of Cationic Copolymerization of Isobutylene and p-(Chloromethyl)styrene[J]. Acta Polymerica Sinica, 2020, 51(4): 355-365. doi: 10.11777/j.issn1000-3304.2019.19179
异丁烯与对氯甲基苯乙烯正离子共聚合研究
摘要:
以四氯化钛(TiCl4)、二氯乙基铝(AlEtCl2)、倍半铝(AlEt1.5Cl1.5)、三氯化铝(AlCl3)等路易斯酸为共引发剂,水或枯基醇(CumOH)为引发剂,在−80 °C下的正己烷/二氯甲烷(V/V = 6/4)的混合溶剂内,研究了异丁烯(IB)与对氯甲基苯乙烯(p-CMS)的正离子共聚合. 利用示差凝胶渗透色谱仪(GPC-RI)以及核磁氢谱(1H-NMR)对共聚物的表观分子量及分子量分布、共聚组成等进行分析,采用Kelen-Tüdős与Yezreielv-Brokhina-Roskin法计算了单体竞聚率,初步探讨了p-CMS与IB正离子共聚合的反应机理. 结果表明,AlEtCl2、AlEt1.5Cl1.5、AlCl3均可催化大分子间的烷基化反应,产生凝胶;TiCl4作为共引发剂,可以得到无凝胶单峰分布共聚物;邻位氯甲基苯乙烯(o-CMS)不能参与共聚,p-CMS的共聚活性较低,IB与p-CMS的单体竞聚率为rIB = 4.67,rp-CMS = 0.70;随反应时间延长,共聚物中p-CMS的含量及共聚物分子量均逐渐增加;p-CMS单体自身几乎不参与引发,共聚到大分子链后,苄基氯缓慢参与引发,形成支化. 提高共聚合温度至− 60和− 40 °C,聚合速率降低,p-CMS的引发活性未发生明显变化.
English
Study of Cationic Copolymerization of Isobutylene and p-(Chloromethyl)styrene
Abstract:
Cationic copolymerization of isobutylene (IB) and chloromethylstyrene was investigated with n-hexane (Hex)/dichloromethane (CH2Cl2) (V/V = 6/4) as solvent, TiCl4, AlEt1.5Cl1.5, AlEt2Cl, AlCl3 as co-initiators and water or cumyl alcohol as initiators. The molecular weight, molecular weight distribution (MWD) and structure composition of the resulting copolymers were analyzed by gel permeation chromatography (GPC) and 1H-NMR spectroscopy. The reactivity ratios were determined by Kelen-Tüdős and Yezreielv-Brokhina-Roskin formula, and the copolymerization mechanism was proposed. It was found that co-initiators with strong Lewis acidity, such as AlEtCl2, AlEt1.5Cl1.5 and AlCl3 can catalyze intermolecular alkylations to form gels while no gel formed with the relatively weaker TiCl4. The chloromethylstyrene with para-substituent, i.e., p-(chloromethyl)styrene (p-CMS) was found to have a low reactivity during the copolymerization with IB (rIB = 4.67, rp-CMS = 0.70) while the ortho-isomer exhibited no activity. The chemical structure of resulting copolymers indicated that p-(chloromethyl)styrene cannot initiate the polymerization of IB which may be due to its low initiation rate compared to the highly active cumyl group at p-CMS/IB molar ratio of 4.11. However, the benzyl chloride group in the formed copolymer chain can slowly initiate polymerization of IB and p-(chloromethyl)styrene, forming branched structures. The content of p-(chloromethyl)styrene increased with increasing molecular weight and monomer conversion. Systematic research on the branched structure, rheological properties and other physical properties of the resulting copolymers is in progress.
-
Key words:
- Isobutylene
- / p-(Chloromethyl)styrene
- / Reactivity ratio
- / Cationic copolymerization
- / Branching
-
卤化丁基橡胶(CIIR/BIIR)具有优异的气密性,广泛应用于无内胎轮胎气密层. 但CIIR/BIIR中的烯丙基卤素在高温下,会发生异构化甚至卤化氢的脱除反应[1];橡胶硫化后,还会残留部分烯丙基双键,这都对耐热性产生不利影响. Exxon公司通过对甲基苯乙烯和IB正离子共聚合及进一步的自由基溴化反应,成功商业化了含高活性苄基溴基团的全饱和橡胶-溴化异丁烯对甲基苯乙烯共聚物(BIMS),该橡胶表现出了更好的耐热性、耐臭氧性、耐候性[2]. 与BIMS结构类似,含苄基氯基团的氯甲基化苯乙烯(CMS)和IB共聚物(CIMS),也同样具备硫化活性[3~7],Exxon 公司将其与尼龙共混,成功制备了气密性更加优异的动态硫化热塑性弹性体[8,9].
目前,文献报道的CIMS的合成方法主要有两类:(1) p-CMS和IB的正离子共聚法直接制备[3~7,10~13];(2)通过IB与苯乙烯(St)正离子共聚合、后氯甲基化顺序合成[14]. 采用路线(1),可以省去繁琐、有毒的氯甲基化反应,具有更高的工业价值. p-CMS为双官能单体,双键可参与共聚,但共聚活性较IB低[3~5],苄基氯也可形成苄基阳离子活性中心[15];大分子链活性中心还可向p-CMS上的苯环发生分子间烷基化,导致凝胶[3~6]. 上述多种反应导致了p-CMS和IB的正离子共聚合基元反应比较复杂,可控性较差,相应的文献报道也比较少.
Jones等[3~5]曾以BF3为共引发剂,在氯乙烷中实现了p-CMS和IB的无凝胶淤浆共聚合. Exxon公司[6,7]认为淤浆聚合易产生凝胶,当采用EDC为共引发剂,在正己烷(n-Hex)和氯甲烷(CH3Cl)的混合溶剂中进行p-CMS和IB的连续法共聚合时,可以抑制凝胶,得到分子量数万的CIMS. Kennedy等[16]也曾经报道过二乙基氯化铝(AlEt2Cl)共引发的p-CMS/IB正离子共聚合,作者认为p-CMS中的苄基氯能够引发,但双键不参与共聚. Nuyken等[10]以三甲基铝为共引发剂,经过仔细研究发现,p-CMS既可以引发,又可以共聚,形成支化. Nuyken[11~13]进一步以四氧化钛(TiCl)4或者BCl3为共引发剂,在二氯甲烷(CH2Cl2)中合成了p-CMS摩尔含量分别高达58%和27%的CIMS,作者侧重于侧链改性,进一步合成了聚异丁烯-聚噁唑烷双亲接枝共聚物. 这些早期文献对p-CMS和IB的正离子共聚合已经进行了许多非常有价值的研究工作,但关于竞聚率、分子结构详细表征、聚合动力学方面的研究数据还比较少,有必要对其进行更深入的研究.
本文在 −80 °C下的n-Hex/CH2Cl2 (V/V = 6/4)混合溶剂内,首先考察了不同路易斯酸对凝胶的影响,并进一步优选TiCl4为共引发剂,研究了p-CMS和IB的共聚合,以Kelen-Tüdős与Yezreielv-Brokhina-Roskin法计算了竞聚率,分析了聚合过程中的一些基元反应,对支化结构进行了证实,初步探讨了聚合机理,为实现直接共聚法合成高分子量CIMS提供了一定的实验基础.
1. 实验部分
1.1 原料
TiCl4 (AR,西亚试剂)、枯基醇(CumOH, 97%,Aldrich)、2,6-二叔丁基吡啶(DtBP,97%,Aldrich)、AlCl3(99%,Aldrich)、二氯乙基铝(AlEtCl2,1 mol/L n-Hex溶液)和二乙基氯化铝(AlEtCl2,1 mol/L n-Hex溶液)均为江苏爱姆欧光电材料有限公司产品、倍半铝(AlEt1.5Cl1.5:AlEtCl2与AlEtCl2以1:1的摩尔比混合制得)、氯乙酰氯(AR,西亚试剂)、无水乙醇(AR,天津富宇化工). 正己烷(n-Hex,AR,国药化学试剂)、二氯甲烷(CH2Cl2,AR,国药化学试剂),高纯氮气保护下在CaH2存在下回流24 h,蒸馏后使用. 异丁烯(IB,聚合级,潍坊普鑫化工有限公司,气体经CaCl2干燥后,冷却成液态使用);对氯甲基苯乙烯(p-CMS,对位与邻位异构体混合物,西亚试剂).
1.2 聚合过程
聚合反应在氮气氛围下的聚合瓶中进行. 典型的聚合过程如下:在n-Hex与CH2Cl2体积比为6:4的混合溶剂中,导入DtBP、CumOH、IB与p-CMS共聚单体,配制单体溶液;然后用导管取22 mL配置好的单体溶液导入氮气保护的聚合瓶内,在−80 °C下的酒精浴中冷却10 min后,用注射器导入TiCl4引发聚合,到达设计时间后,加入预冷乙醇终止反应. 聚合物经过多次n-Hex/乙醇沉淀提纯,40 °C真空干燥至恒重.
1.3 测试与表征
1.3.1 p-CMS共聚含量与苄基氯含量以及支化程度的计算
根据参考文献[15,17~20], p-CMS中(―CH2Cl)亚甲基质子的吸收峰在δ = 4.56处;苄基氯引发IB聚合后,亚甲基质子(―CH2―)的吸收峰在δ = 2.75;PIB主链亚甲基质子吸收峰在δ = 1.41. 定义ICMS、
$F_{\rm CH_2Cl} $ $ {I_{{\rm{CMS}}}} = \dfrac{{\dfrac{{{A_{4.56}} + {A_{2.75}}}}{2}}}{{\dfrac{{{A_{4.56}} + {A_{2.75}}}}{2} + \dfrac{{{A_{1.41}}}}{2}}} $ (1) $ {F_{{\rm{C}}{{\rm{H}}_2}{\rm{Cl}}}} = \dfrac{{\dfrac{{{A_{4.56}}}}{2}}}{{\dfrac{{{A_{4.56}} + {A_{2.75}}}}{2} + \dfrac{{{A_{1.41}}}}{2}}} $ (2) 其中A4.56、A2.75、A1.41表示在核磁谱图中相应峰的峰面积.
苄基引发点代表了分子链中的支化点,根据GPC-RI的表观数均分子量数据(Mn),以支化点在共聚物中的摩尔比与单体聚合度的积,计算了平均每条分子链中的支化点(S)的数目,具体计算公式为:
$ {{S}} = \frac{{{M_{\rm{n}}}}}{{56 \times \left( {1 - {I_{{\rm{CMS}}}}} \right) + 152 \times {I_{{\rm{CMS}}}}}} \times \left( {{I_{{\rm{CMS}}}} - {F_{{\rm{C}}{{\rm{H}}_2}{\rm{Cl}}}}} \right) $ (3) 1.3.2 测试仪器
1H-NMR在型号为布鲁克500UltraShield核磁共振仪上测试,测试溶剂为氘代氯仿,四甲基硅烷(TMS)为标准参照物,将10.0 mg左右聚合物样品完全溶解于核磁样品管中;GPC-RI数据采用日本TOSOH株式会社型号EcoSEC8320的设备测试,柱温40 °C,以四氢呋喃为流动相,流速0.35 mL/min. 采用Wyatt公司示差折光指数仪(RI)/多角激光光散射仪(LS) /在线黏度检测器(Vis) 三台联用设备测定Mark-Houwink-Sakurada方程中的K, α值,流动相为THF,流速1.0 mL/min,柱温25 °C. 示差扫描量热仪(DSC)为瑞士梅特勒托利多仪器制造有限公司生产的DSC1型,测试温度为−120 ~ 30 °C,氮气氛围,升温速率为10 °C/min.
2. 结果与讨论
2.1 引发体系
首先,以CumOH/TiCl4、H2O/AlEtCl2、H2O/AlEt1.5Cl1.5、H2O/AlEt2Cl、H2O/AlCl3为引发体系,在n-Hex/CH2Cl2 (V/V = 6/4)的混合溶剂中引发CMS和IB共聚合. 实验现象如表1所示,H2O/AlEt2Cl引发体系没有聚合活性;H2O/AlEtCl2、H2O/AlEt1.5Cl1.5引发体系的聚合速度很快,体系从无色迅速转变为亮红色,同时有大量红色不溶物析出;苯甲醚作为给电子体时,H2O/AlCl3引发体系无聚合活性;氯乙酰氯为给电子体时,聚合速度很快,迅速产生红色凝胶;只有CumOH/TiCl4引发体系,聚合平稳,体系呈现浅黄色. 这表明,AlEt1.5Cl1.5和AlEtCl2的路易斯酸酸性太强,极易催化活性中心向苯环的分子间烷基化反应,产生凝胶. 当选用给电子性相对较强的苯甲醚时,AlCl3无聚合活性,而选用氯乙酰氯作为给电子体时,2个氯原子的吸电子诱导作用,降低了氯乙酰氯的给电子能力,此时AlCl3的路易斯酸性强,分子间烷基化催化活性很高,体系很容易产生凝胶. 因此,优选TiCl4做进一步的研究.
表 1

 下载:
导出CSV
下载:
导出CSV
Initiator Co-initiator Electron donor Phenomenon CumOH TiCl4 − Polymer; no gel; pale yellow H2O AlEtCl2 − Polymer and gel; red H2O AlEt1.5Cl1.5 − Polymer and gel; red H2O AlEt2Cl − No polymer H2O AlCl3 Anisole No polymer H2O AlCl3 Chloroacetyl chloride Polymer and gel; red Reaction conditions: [co-initiator] = 0.045 mol/L, [CumOH] = 0.01 mol/L, ED/AlCl3 (molar ratio) = 1.1/1, n-Hex/CH2Cl2 = 6/4 (V/V), [IB] = 1.63 mol/L, [CMS] = 0.33 mol/L, T = − 80 °C, reaction time = 30 min 2.2 竞聚率
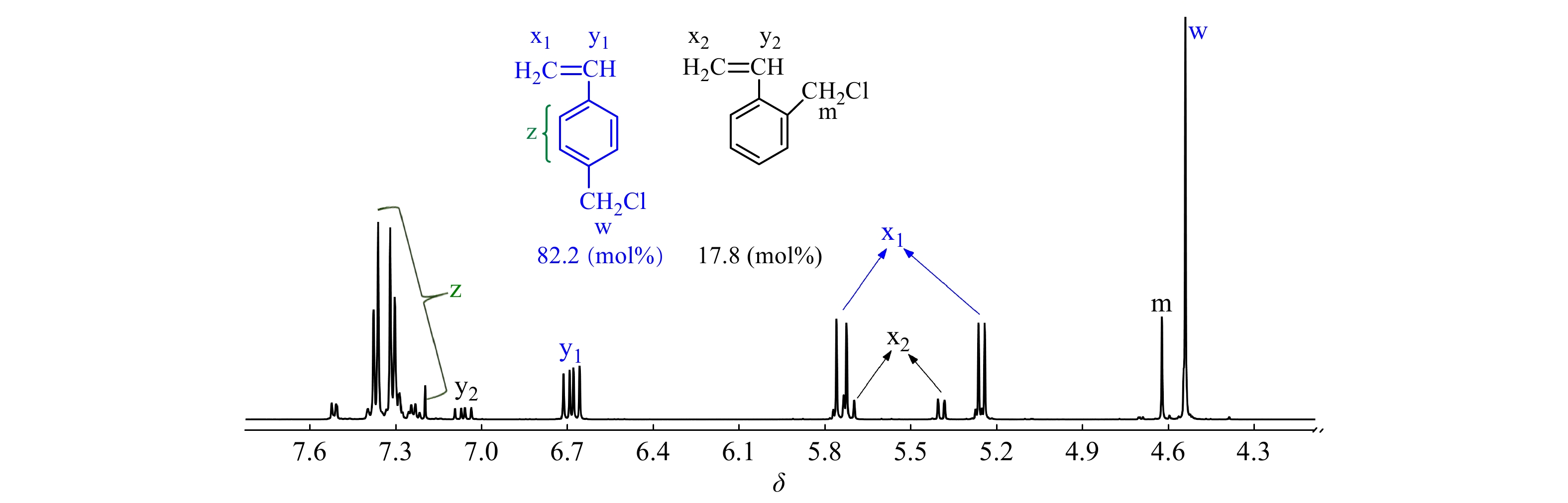
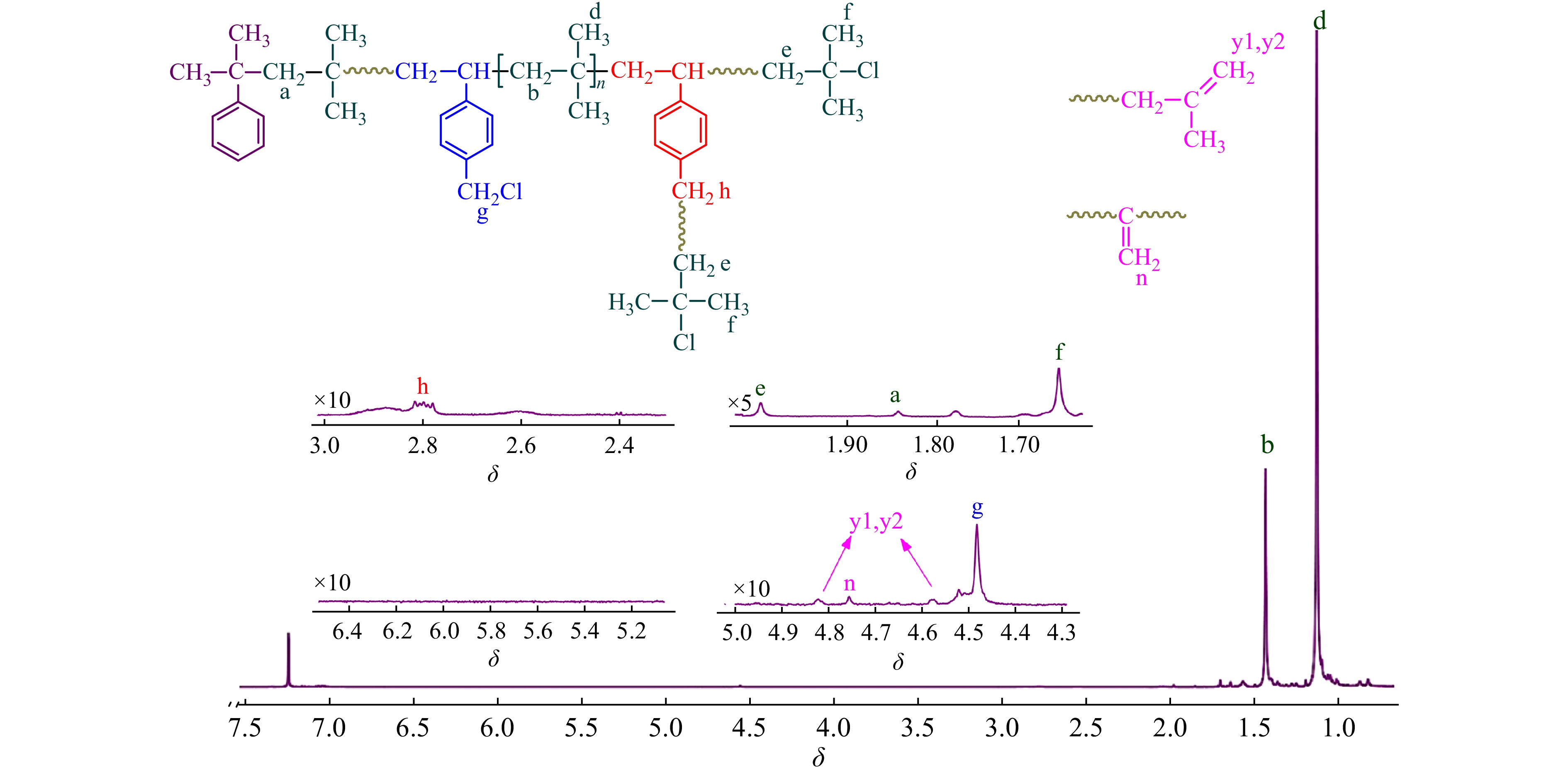
现有文献尚鲜有以TiCl4为共引发剂时CMS和IB的竞聚率数据报道. 以CMS代表邻位和对位氯甲基化苯乙烯异构体混合物,首先在CMS/IB = 2.5/100 ~ 50/10(摩尔比)的范围内进行共聚合,控制单体转化率在15%以下,以计算竞聚率. 图1为所用CMS单体的1H-NMR谱图,p-CMS的苄基质子吸收峰在δ = 4.54,o-CMS的苄基质子吸收峰在δ = 4.62,经计算,p-CMS/o-CMS的摩尔比为82.2/17.8.
图 1
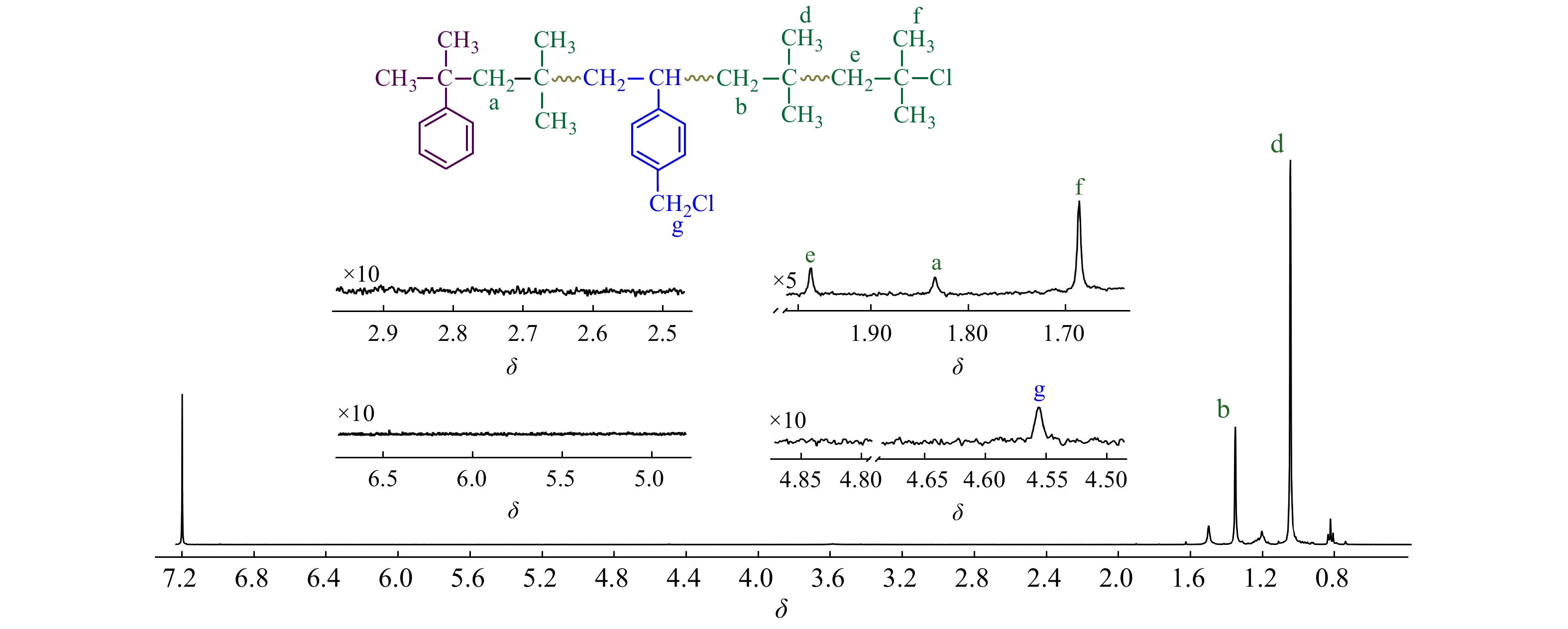
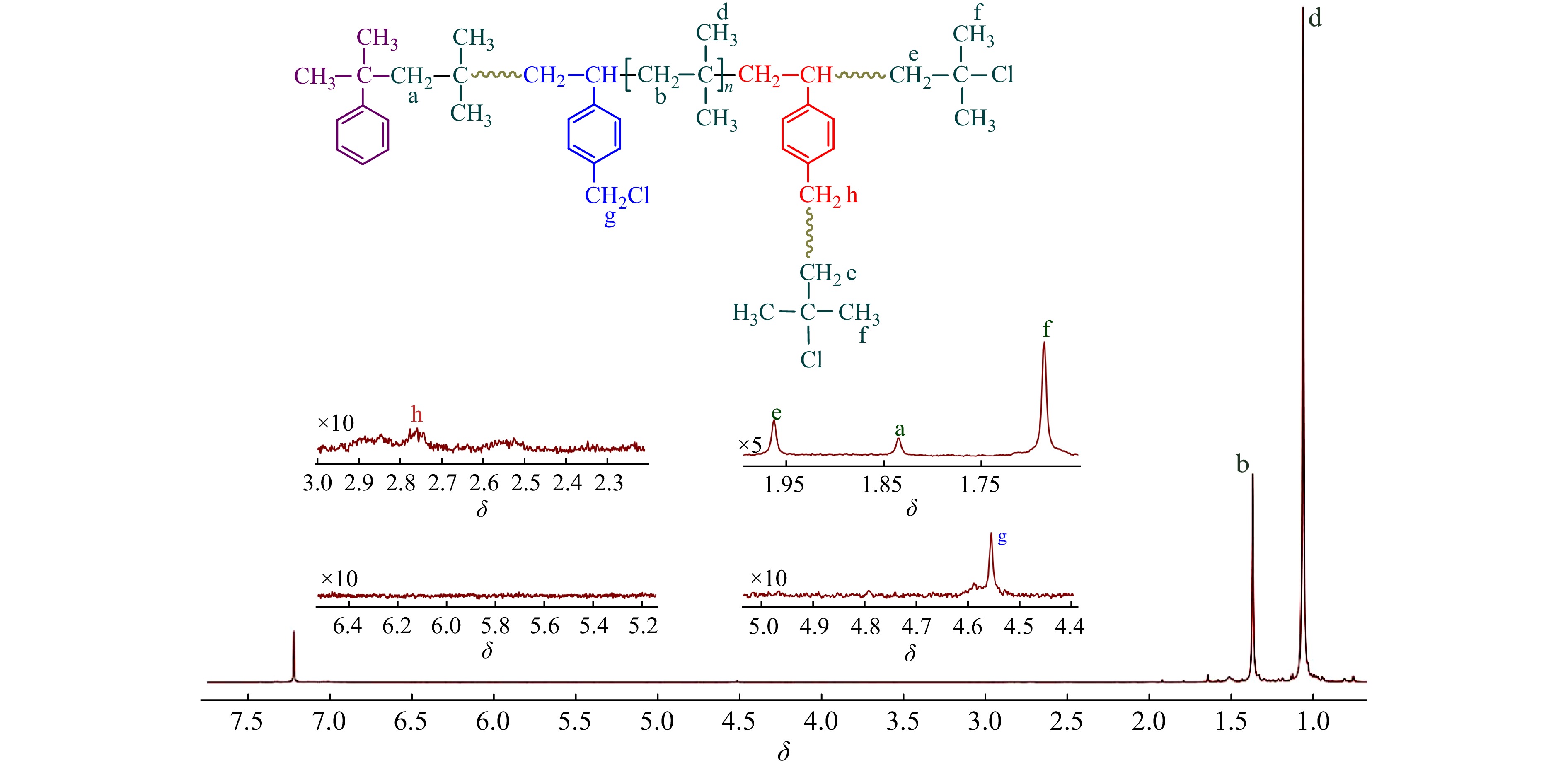
图2为CMS/IB的摩尔比为2.5/100时共聚产物的1H-NMR谱图,可清晰地观察到[17,18]:δ = 1.41处为共聚物IB单元亚甲基质子吸收峰;δ = 1.11处为IB单元侧甲基质子吸收峰;δ = 1.84处为枯基所引发的首个IB单体的亚甲基质子吸收峰;δ = 4.56处为苄氯亚甲基质子吸收峰;δ = 1.96和δ = 1.68 处为末端叔丁基氯特征峰;在δ = 4.64/4.85/5.15处未观察到PIB末端双键的特征吸收峰,表明共聚体系内不存在末端质子脱除等副反应. 从图2中还可以看到,在δ = 4.56处仅观察到一个单峰,对应p-CMS单体,这表明o-CMS无法与IB进行正离子共聚,真正有效参与共聚的单体为p-CMS.
图 2
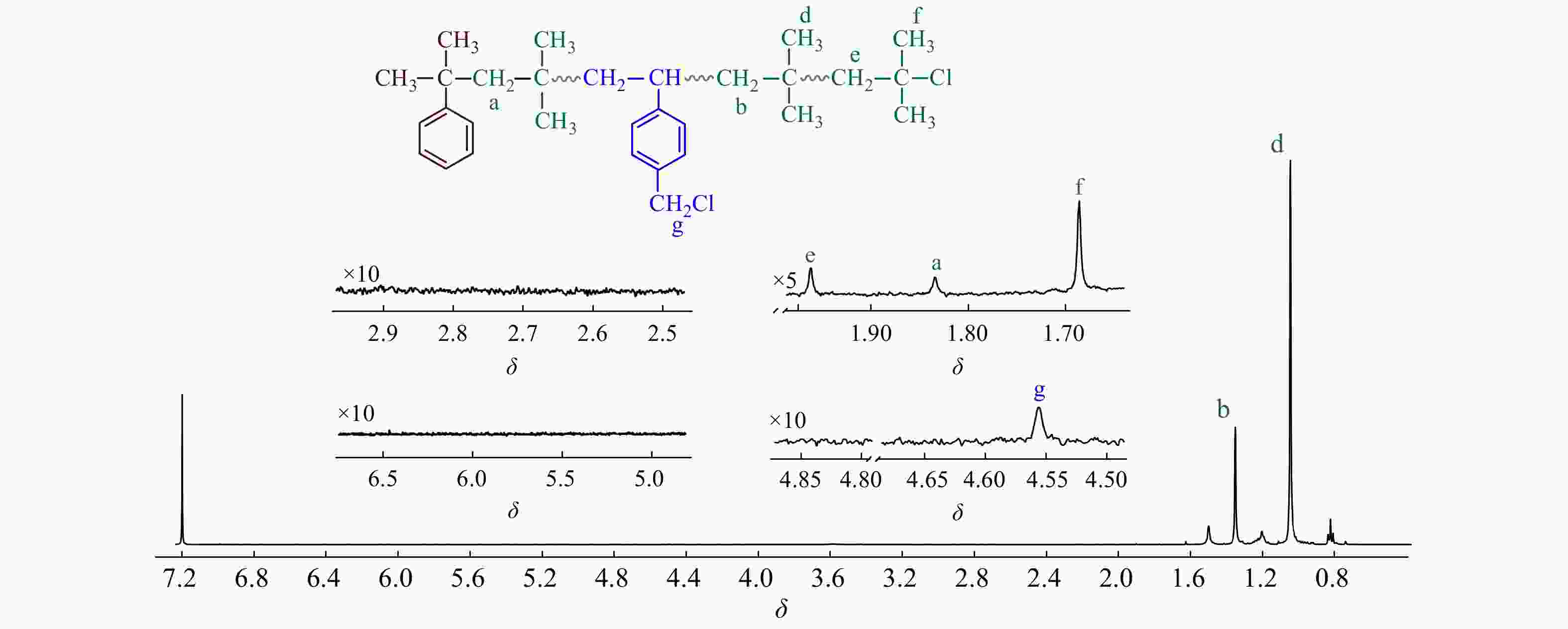 Figure 2. 1H-NMR spectrum of the resulting copolymer (Conditions: CMS/IB = 2.5/100 (molar ratio). [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L, reaction time = 6 min, T = −80 °C)
Figure 2. 1H-NMR spectrum of the resulting copolymer (Conditions: CMS/IB = 2.5/100 (molar ratio). [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L, reaction time = 6 min, T = −80 °C)p-CMS单体中的苄氯可以2种形式参与引发:(1)单体自身作为引发剂,引发聚合,形成端乙烯基的大分子单体;(2)共聚进入大分子链后,在侧链引发聚合. 但在图2中,δ = 2.75 处未观察到苄基引发特征吸收峰[15,19~20],δ = 5.20和5.70附近也未观察到苯乙烯基双键的吸收峰[20]. 这表明,苄基的引发活性远低于枯基[21],当单体转化率较低、反应时间较短时,共聚进入分子链中的苄基氯未起到明显的引发作用.
单体共聚的竞聚率是指单体在共聚时进入分子链的能力,计算方法主要有3种:Fineman Ross (F-R)法、Kelen-Tüdős (K-T)法、Yezreielv-Brokhina-Roskin (YBR)法[21~23]. 3种方法都是基于单体初始投料比与在聚合物中的共聚比进行计算. F-R方法计算方便但是结果准确性不高[21~23],因此本文采用K-T法与YBR法计算竞聚率. 表2给出了不同p-CMS/IB投料比下单体转化率与p-CMS共聚含量. 图3为通过K-T方法所计算的正序值与逆序值所绘制的回归线图(a为正序、b为逆序). YBR法则是通过一系列数学处理方法来计算得到竞聚率,经计算,rIB = 4.68,误差1.6%,rp-CMS = 0.69,误差3.0%. 这表明rIB 和rp-CMS 具有较高的可信度. 最终通过2种方法求得的平均值如表3所示,rIB = 4.67,rp-CMS = 0.70.
表 2
Table 2. Copolymer compositions by 1H-NMR下载:
导出CSV
Run Conversion (%) Monomer feeding (%) Copolymer composition (%) IB p-CMS IB p-CMS − 100 0 100 0 1 11.4 97.88 2.12 99.54 0.46 2 9.5 91.65 8.35 97.96 2.04 3 13.9 82.95 17.05 95.36 4.64 4 8.5 73..95 26.05 90.94 9.06 5 7.56 64.60 35.40 88.13 11.87 6 7.3 54.89 45.11 82.02 17.98 − 0 100 0 100 Conditions: [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L, reaction time = 6 min, T = −80 °C 图 3
表 3
Table 3. sReactivity ratios of the p-CMS-IB copolymerization systems下载:
导出CSV
Methods Positive sequence Reverse rIB rp-CMS rIB′ rp-CMS′ K-T 4.658 0.7195 4.6582 0.7196 YBR 4.676 ± 0.075671 0.6886 ± 0.02088 Average rIB = 4.67 rp-CMS = 0.70 2.3 共聚合时间
2.3.1 共聚合动力学
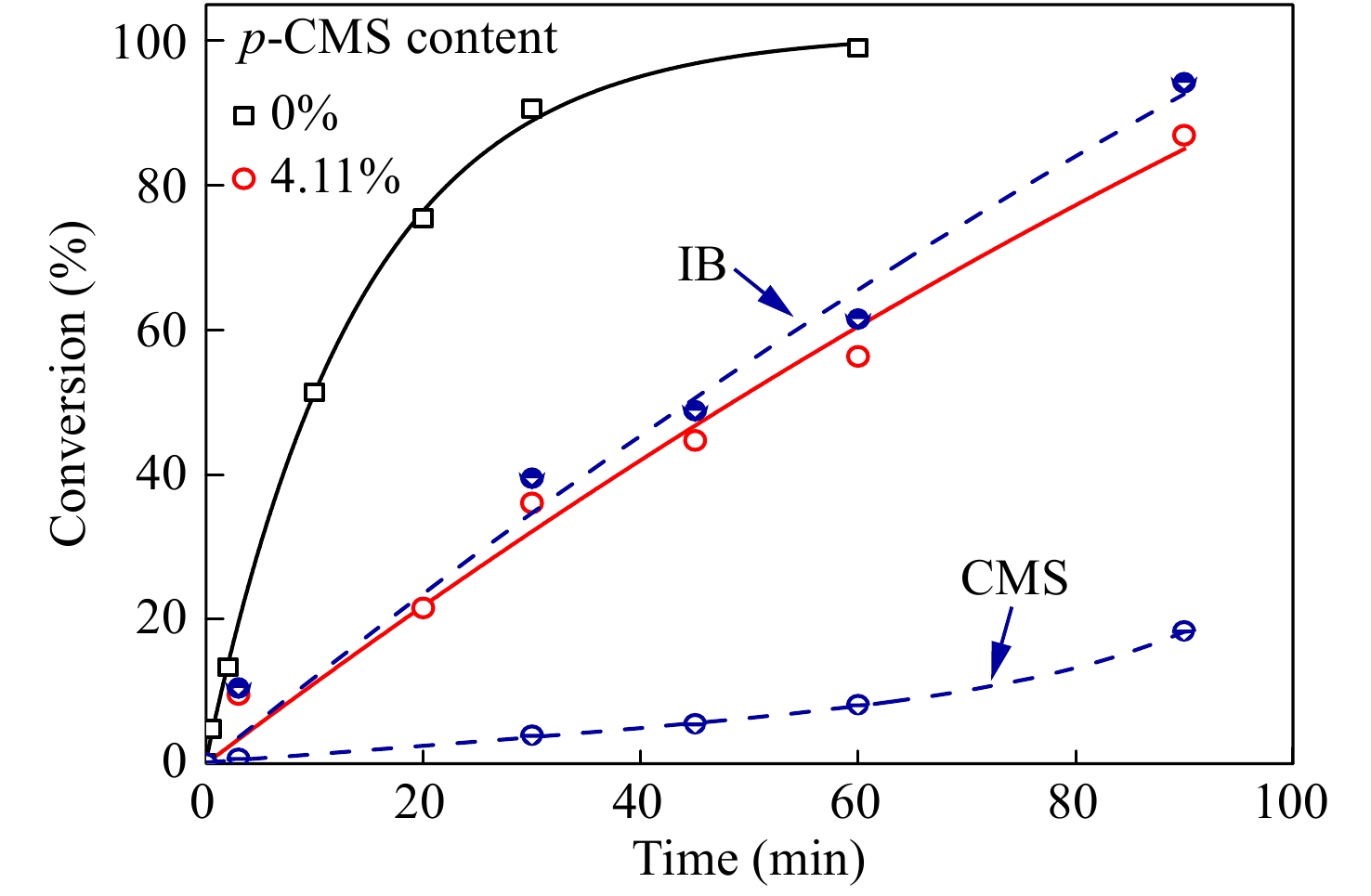
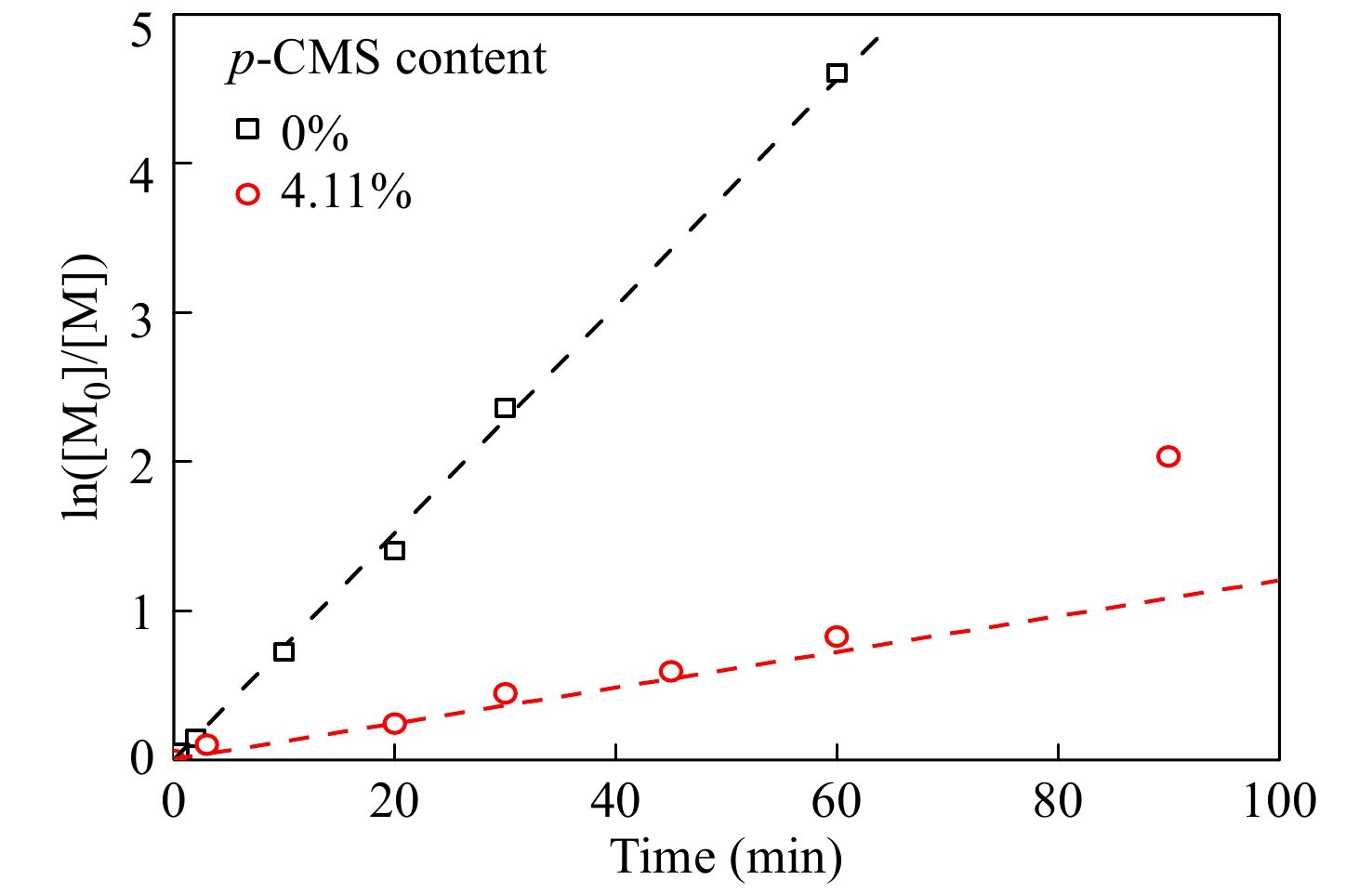
进一步在p-CMS/IB的摩尔比为4.11/100的条件下,研究共聚合时间的影响,并与IB均聚合速率作对比. 由图4共聚时间与单体转化率曲线可知,引入少量p-CMS后,整体聚合速率有所降低,但聚合反应仍能平稳进行. ln([M0]/[M])动力学曲线如图5所示,在反应时间45 min以内时,共聚速度与单体浓度呈现一级动力学关系,但在45 min以后,共聚速度与单体浓度偏离一级动力学关系,聚合速度加快,表明体系内活性中心的浓度明显增加.
图 4
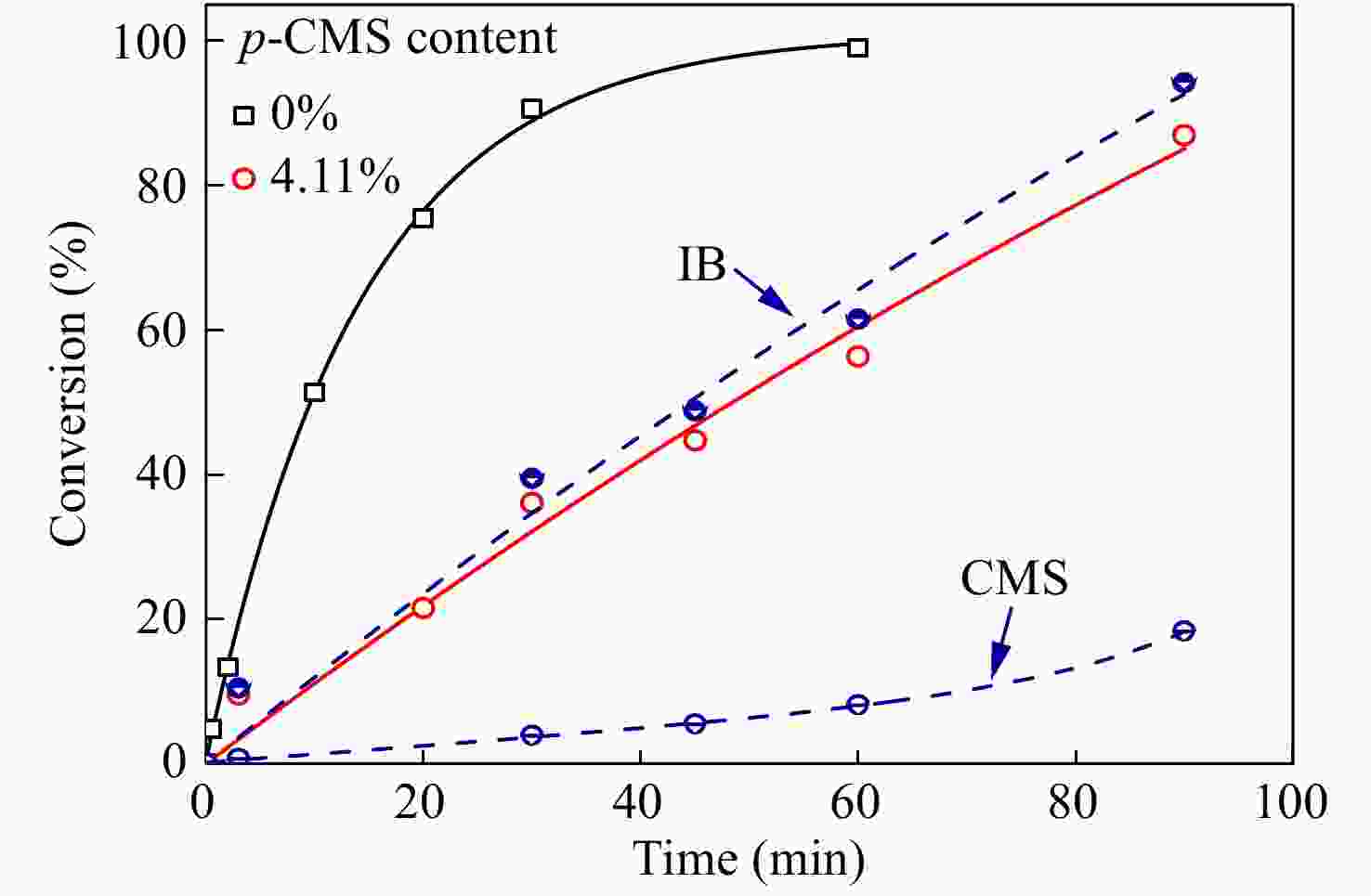 Figure 4. Plots of conversion versus time for p-CMS-IB copolymerization (Conditions: [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L, T = −80 °C)
Figure 4. Plots of conversion versus time for p-CMS-IB copolymerization (Conditions: [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L, T = −80 °C)图 5
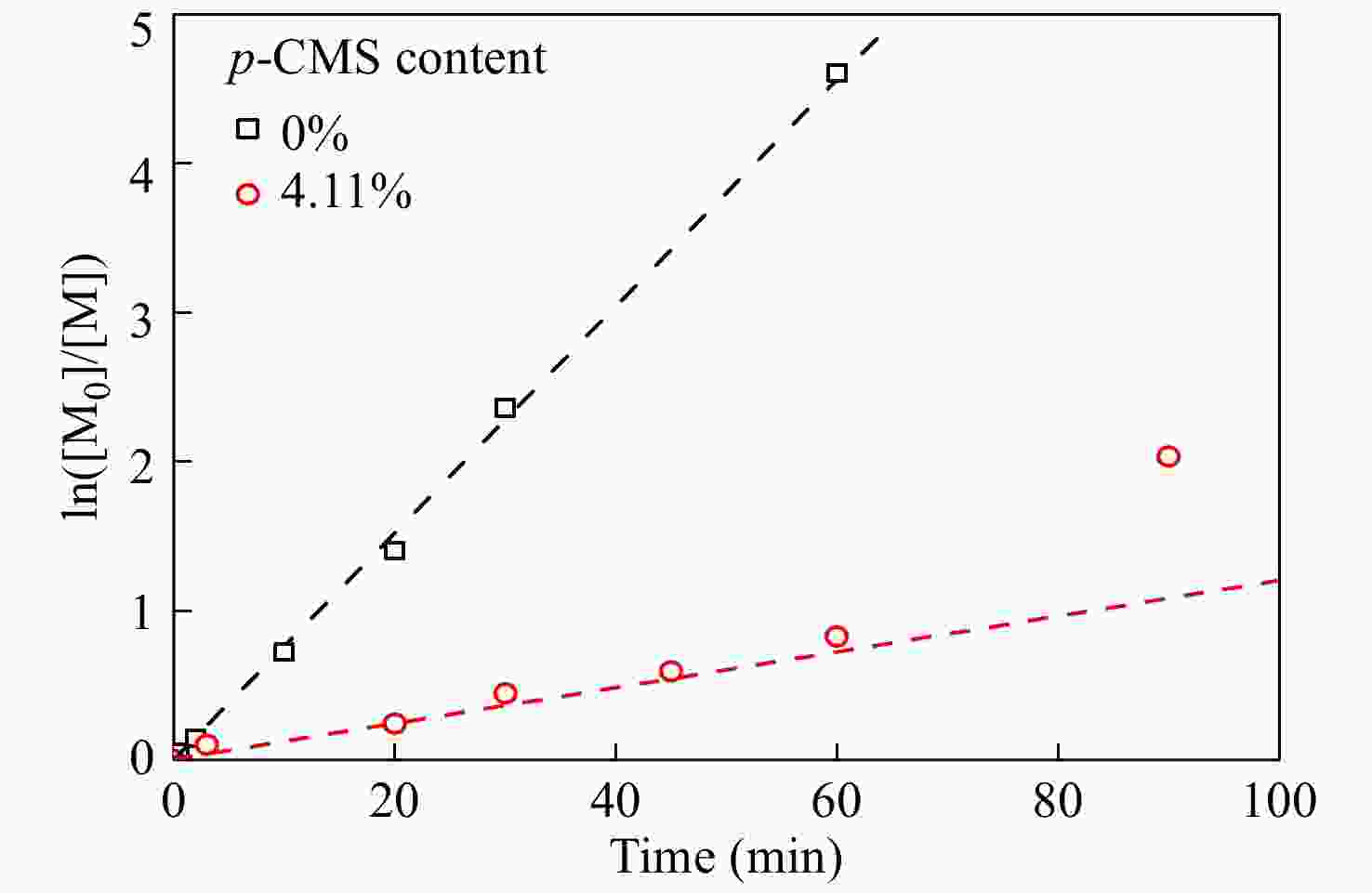 Figure 5. Plots of first-order plots of ln([M0]/[M]) versus time for p-CMS-IB copolymerization
Figure 5. Plots of first-order plots of ln([M0]/[M]) versus time for p-CMS-IB copolymerization2.3.2 GPC-RI 表征
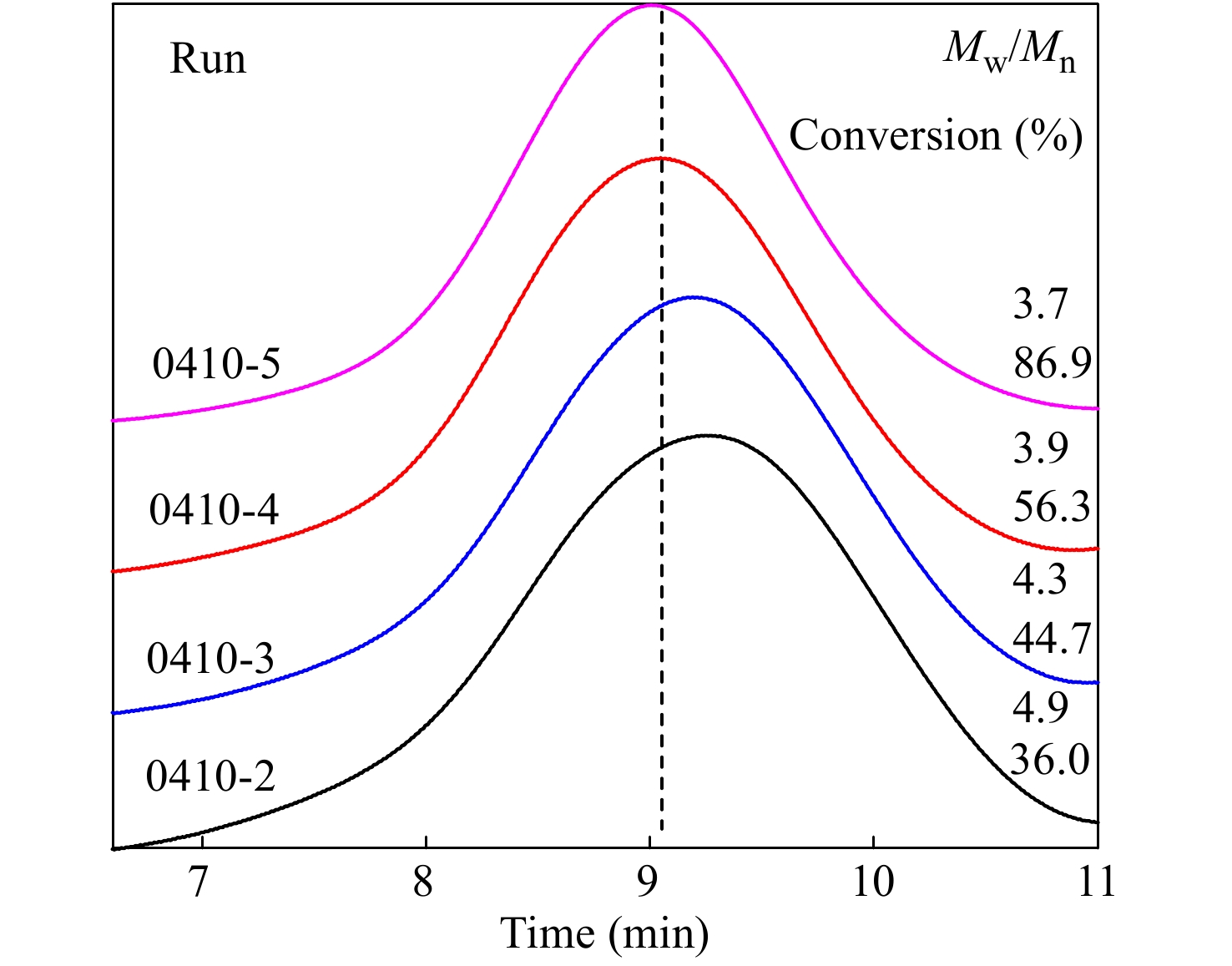
对转化率较高的共聚产物进行了GPC-IR表征,结果见图6及表4所示,共聚物的分子量分布较宽,分子量分布指数(MWD, Mw/Mn)达到4左右,但均呈单峰分布. 如前节所述,在p-CMS和IB共聚体系内,不存在末端质子脱除等副反应,大分子链末端基为有活性的叔丁基氯结构. 如图6所示,随反应时间延长和单体转化率的提高,共聚物的表观数均分子量和峰位分子量不断提高,这也证实了大分子链末端基具有活性.
表 4
Table 4. The 1H-NMR data of the resulting copolymers下载:
导出CSV
Run Reaction time
(min)Mn,GPC,RI $F_{\rm CH_2 Cl} $ 

(mol%)ICMS
(mol%)Branching points
(per chain)BSB
(mol%)SSB
(mol%)0410-1 3 NA 0.69 0.69 0 100 0 0410-2 30 11700 0.8 1.08 0.58 100 0 0410-3 45 12680 0.82 1.23 0.93 72.46 27.53 0410-4 60 15670 0.76 1.45 1.93 70.92 29.08 0410-5 90 16220 0.89 2.13 3.59 59.17 40.83 图 6
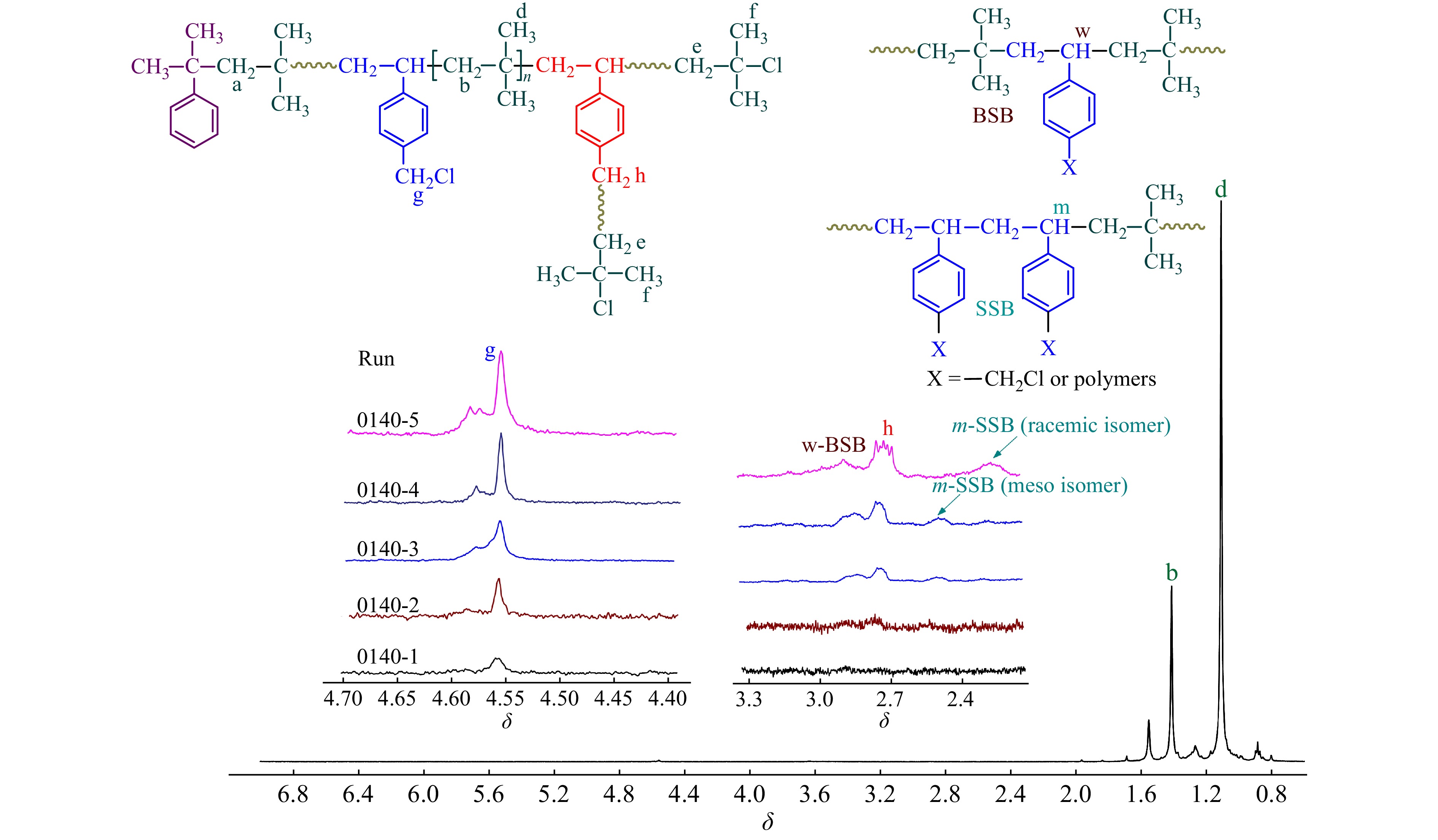
2.3.3 1H-NMR表征
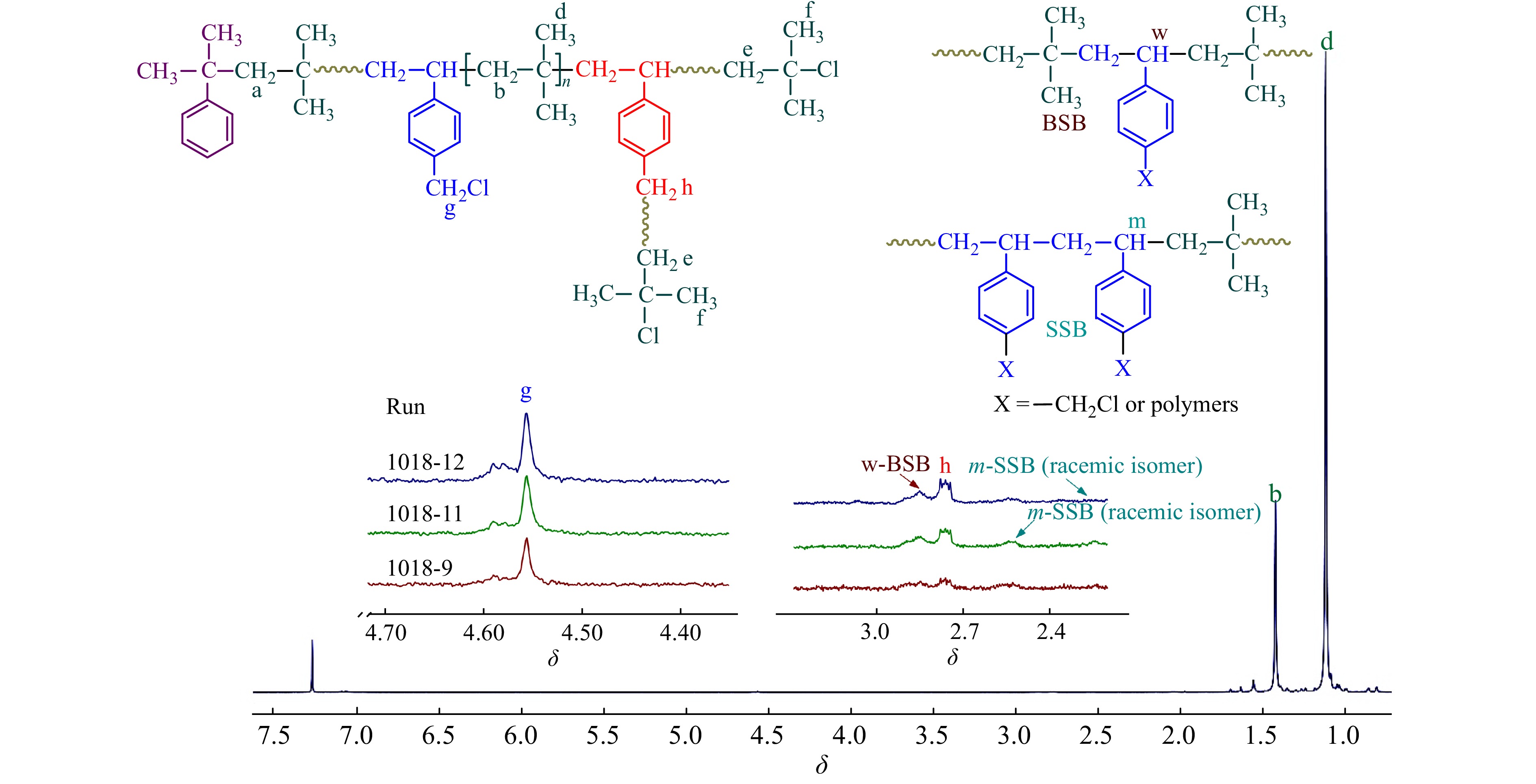
对共聚产物进行了1H-NMR表征,结果如图7所示. 可以看出:(1)随单体转化率的提升,δ = 4.56处苄基氯特征峰逐渐加强;(2) δ = 2.75处苄基氯引发特征峰也开始出现,并不断增加. 根据1H-NMR表征结果,在图4中还绘制了共聚合过程中,IB与p-CMS各自的单体转化率曲线. 从图4可以看出,IB单体的聚合速度要比p-CMS快,这对应了二者的竞聚率数据;随着共聚合时间的延长,p-CMS的浓度相对于IB的浓度不断提升,因此p-CMS在分子链中的含量逐渐增加,且增加的趋势越来越快.
图 7
相比于PIB末端叔丁基氯活性中心,p-CMS中的苄基氯为慢引发[24],在反应初期苄基氯不参与引发,共聚物为线性结构;随反应时间延长,共聚物中苄基氯的引发机率也不断提高,开始形成苄基活性中心引发聚合,形成支化,δ = 2.75处苄基氯引发特征峰强度逐渐增加. 需要特别注意的是,在不同时间下的1H-NMR核磁谱图中,均未观察到明显的苯乙烯双键残留峰,这进一步表明,p-CMS单体几乎不参与直接引发.
通过1H-NMR还可以研究p-CMS在共聚物中的序列分布. 从图7中可以看出,由于两者共聚能力相差较大,在聚合初期,p-CMS的共聚量较少,多以无规共聚(BSB)形式存在;随共聚含量的增加,δ = 2.55和2.3处SSB特征吸收峰出现,并不断增加[25].
表4给出了不同时间下,p-CMS共聚含量、苄基氯残留量以及不同序列结构含量的计算结果. 可以发现,在聚合初期,苄基氯含量达到0.69%,其残留率相对较高,接近BIMS (Exxpro 3433牌号)的苄基官能团的含量;随着时间延长,p-CMS共聚含量逐渐增加,分子链中的苄基氯参与引发,形成支化,整体苄基氯含量变化不大.
2.4 共聚合温度
为了更全面地考察p-CMS的引发活性,在−60和−40 °C下,进行了IB与p-CMS的共聚合. 表5给出了单体转化率以及产物的核磁表征结果. 可以看出,提高聚合温度,明显降低了共聚合速率,在−40 °C下,反应时间90 min时,单体转化率仅达到23%. 图8和图9分别为相近转化率时,−40和−60 °C条件下共聚物的核磁谱图. 从图8和图9中可以清晰地观察到以下特征峰:δ = 1.68和1.96 处叔丁基氯吸收峰;δ = 4.56 处苄氯亚甲基质子吸收峰;δ = 2.75处苄氯引发特征峰等. 另外,在δ = 5.2和5.7处未观察到乙烯基残留峰,表明与−80 °C下的现象一致,p-CMS单体中的苄基氯在−60和−40 °C下,同样不能引发共聚合反应.
表 5
Table 5. The monomer conversion and 1H-NMR data of the resulting copolymers下载:
导出CSV
Run Reaction
time (min)Reaction
temp. (°C)Conversion
(%)$F_{{\rm CH}_2 {\rm Cl}} $ 

(mol%)ICMS
(mol%)BSB
(mol%)SSB
(mol%)1018-4 45 −40 9.62 NA NA NA NA 1018-6 90 −40 23.08 0.88 1.61 74.00 32.00 1018-8 20 −60 15.31 NA NA NA NA 1018-9 30 −60 20.19 0.66 0.99 59.49 40.51 1018-10 45 −60 26.12 NA NA NA NA 1018-11 60 −60 39.42 0.89 1.33 56.59 43.41 1018-12 90 −60 51.92 0.71 1.6 71.83 28.17 Conditions: CMS/IB = 5/100. [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L 图 8
图 9
与图2以及图9相比,在图8 δ = 4.64/4.85,以及δ = 4.79处观察到了少量PIB末端双键以及PIB活性末端双键发生异构化形成的内乙叉基特征吸收峰[18]. 末端双键和内乙叉基与叔丁基氯的摩尔比值为15%. 这表明−40 °C时,共聚体系内发生部分末端质子脱除副反应,使部分端基丧失聚合活性. Faust等[26]研究发现,TiCl4共引发的IB正离子均聚合,在−40 °C时,会出现末端质子脱除副反应,这与本文的研究结果相吻合.
图10为−60 °C时,不同时间下共聚产物的核磁谱图. 从图10及表5中可得到与−80 °C时非常近似的结果:(1)在聚合过程中,随反应时间延长,共聚进入大分子链的p-CMS中的苄基氯不断参与引发,形成支化;(2) p-CMS多以BSB无规共聚形式参与共聚;(3)共聚物中残留苄基氯的含量在0.7% ~ 0.8%左右.
图 10
从温度实验结果看,提高聚合温度,明显降低共聚合速率,但对p-CMS的引发活性影响不明显. 因此,在进一步的研究工作中,将优选较低的聚合温度,以提高聚合速度,减少链末端质子脱除等副反应,提高共聚物分子量.
2.5 支化度
基于GPC-RI和1H-NMR数据,计算了平均每个分子链中的支化点数目,结果如表4所示. 随反应时间延长,共聚物支化点变多,支化程度不断提高. 这些聚合特征,表明TiCl4共引发的p-CMS与IB的正离子共聚合,属于自缩合乙烯基共聚合(SCVCP)聚合[27,28]:p-CMS作为inimer单体,既可以参与共聚,又可以引发聚合. 与大部分ATRP型[28]以及RAFT型[29,30]SCVCP聚合机理不同,由于苄基氯的慢引发,inimer单体p-CMS的单体自身引发现象不明显.
从表4还可以看出,当聚合时间小于45 min时,平均每个分子链中的支化点在1个以下,支化程度很低,接近于线性聚合物. 反应后期,共聚产物的支化程度不断上升,反应时间延长到90 min后,平均每分子链中的支化点达到3.59个. 显然,此时采用示差检测器,已不能够真实反映聚合物的实际分子量.
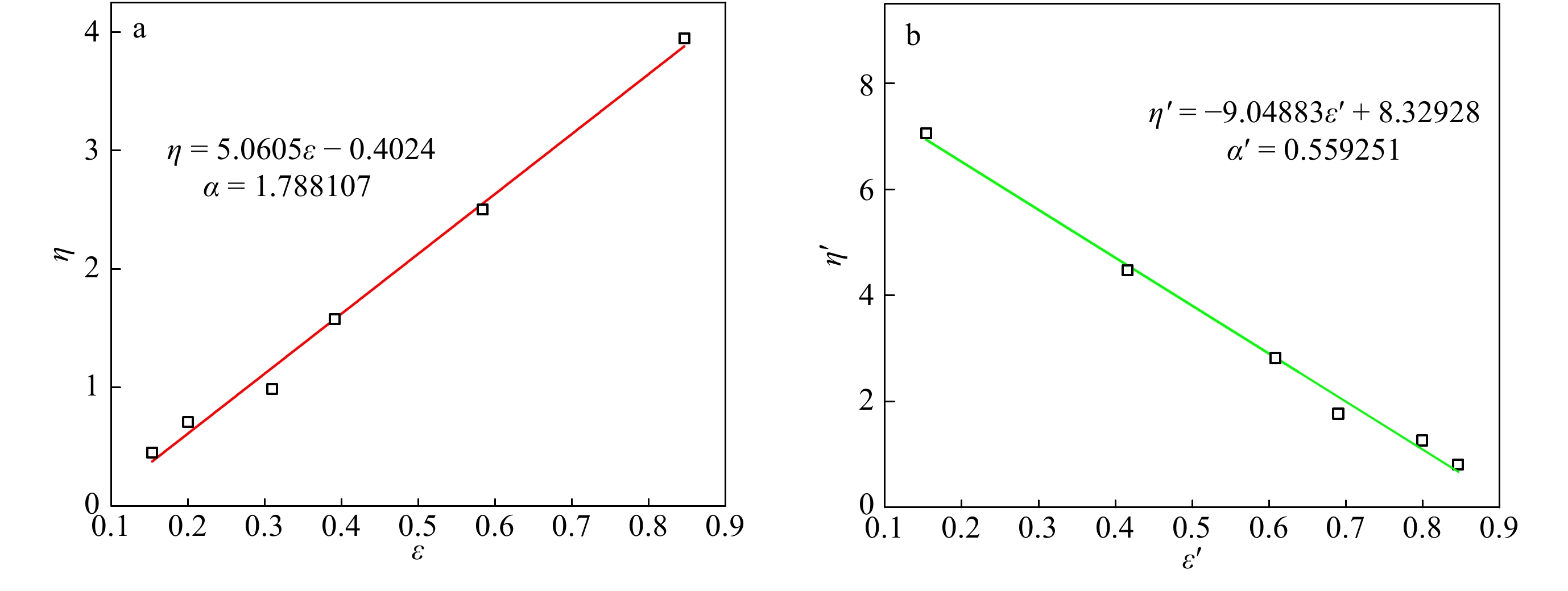
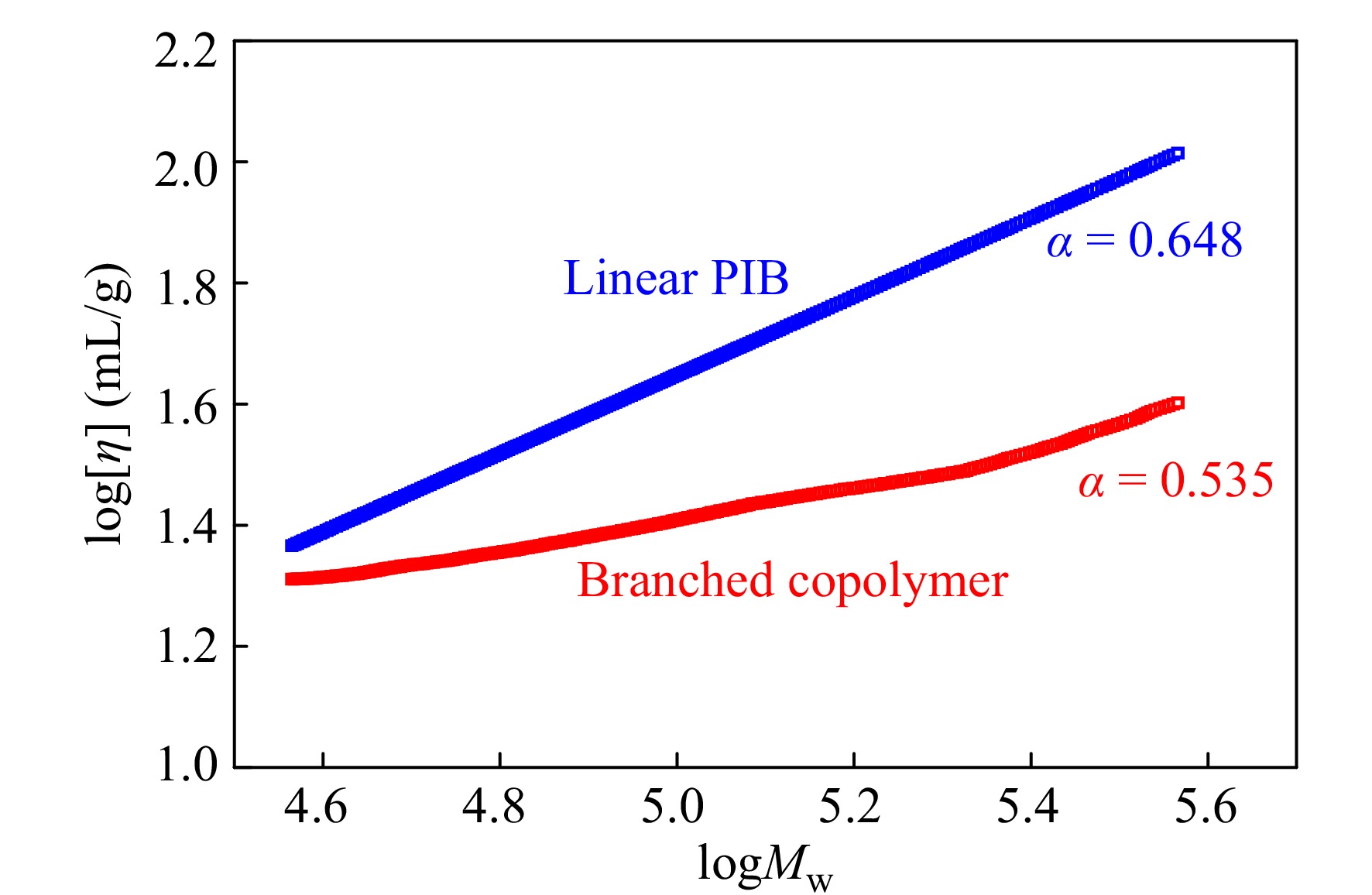
RI/LS/Vis三检测GPC可以通过多个检测器联用,得到绝对重均分子量、均方根旋转半径、特性黏度等数据. 通过特性黏度与绝对重均分子量之间的对数关系曲线,可以得到Mark-Houwink-Sakurada方程中的K, α值. 由于支化聚合物分子密度高,在同等分子量下,相比于线性聚合物具有更小的尺寸和更低的黏度. 因此,支化聚合物的α值会比线性聚合物的低. 图11给出了样品0410-5三检测GPC的测试结果,并与同样条件下TiCl4共引发得到的线性PIB作对比. 可以看出,光散射法测试的0410-5样品的绝对重均分子量明显大于GPC-RI的结果;线性PIB的α值为0.648,而共聚物样品的α值为0.535,这直观地证实了支化结构的存在.
图 11
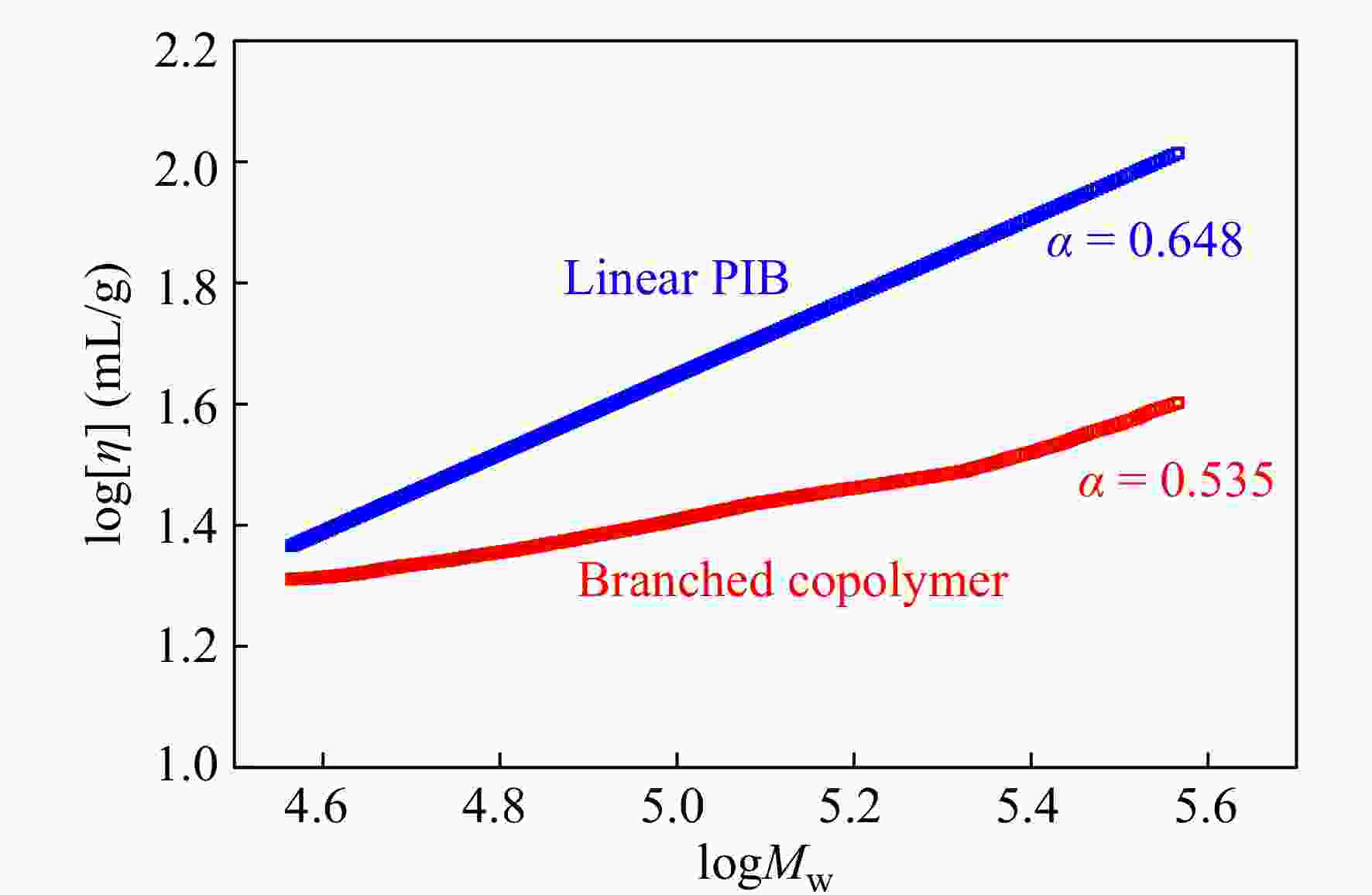 Figure 11. Mark-Houwink-Sakurada plots for linear PIB and IB/p-CMS copolymer (run 0410-5 in Table 4) (LS data: Linear PIB, Mn = 3.9 × 104, Mw = 8.6 × 104, MWD = 2.19; Copolymer, Mn = 5.3 × 104, Mw = 12.5 × 104, MWD = 2.37)
Figure 11. Mark-Houwink-Sakurada plots for linear PIB and IB/p-CMS copolymer (run 0410-5 in Table 4) (LS data: Linear PIB, Mn = 3.9 × 104, Mw = 8.6 × 104, MWD = 2.19; Copolymer, Mn = 5.3 × 104, Mw = 12.5 × 104, MWD = 2.37)2.6 DSC表征
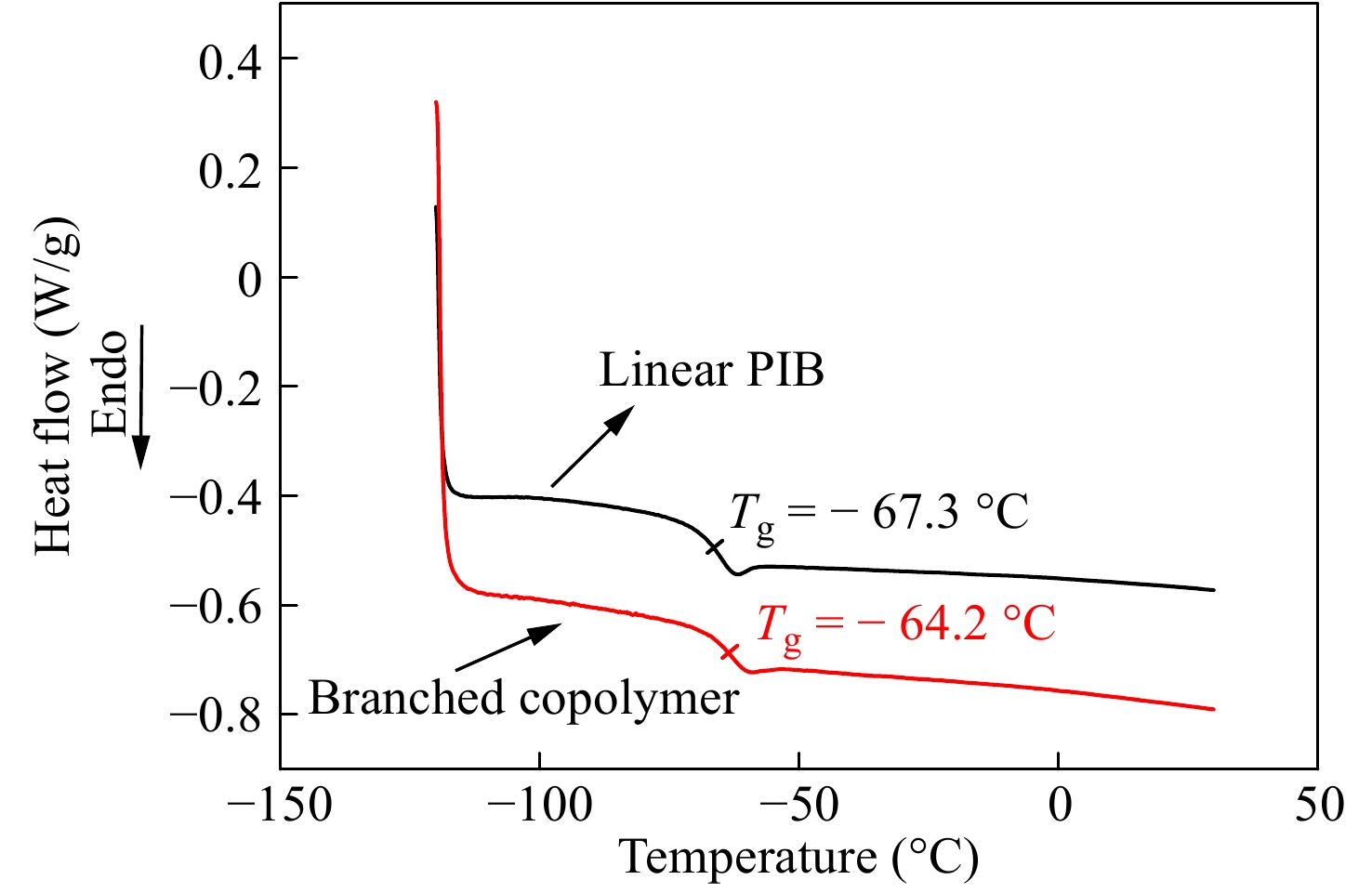
对产物还进行了DSC热分析,以观察支化对玻璃化转变温度(Tg)的影响. 图12为支化样品0410-4以及线性PIB的DSC曲线. 线性PIB的Tg = −67.3 °C,支化样品的Tg = −64.2 °C. Kennedy等[31]研究发现,IB与刚性单体苯乙烯共聚,会提高线性共聚物的玻璃化转变温度,基于PIB和PS的Tg,作者给出了理论与实验值吻合性良好的计算公式. 文献[32]给出聚对氯甲基苯乙烯的Tg = 105 °C,结合文献[31],可知未支化线性共聚物的理论Tg = −65.5 °C,这与实际测试结果非常接近. 这表明少量共聚及轻微支化对Tg影响不大,共聚物仍具有非常低的Tg,这非常有助于其在轮胎气密层中的应用.
图 12
2.7 共聚合机理
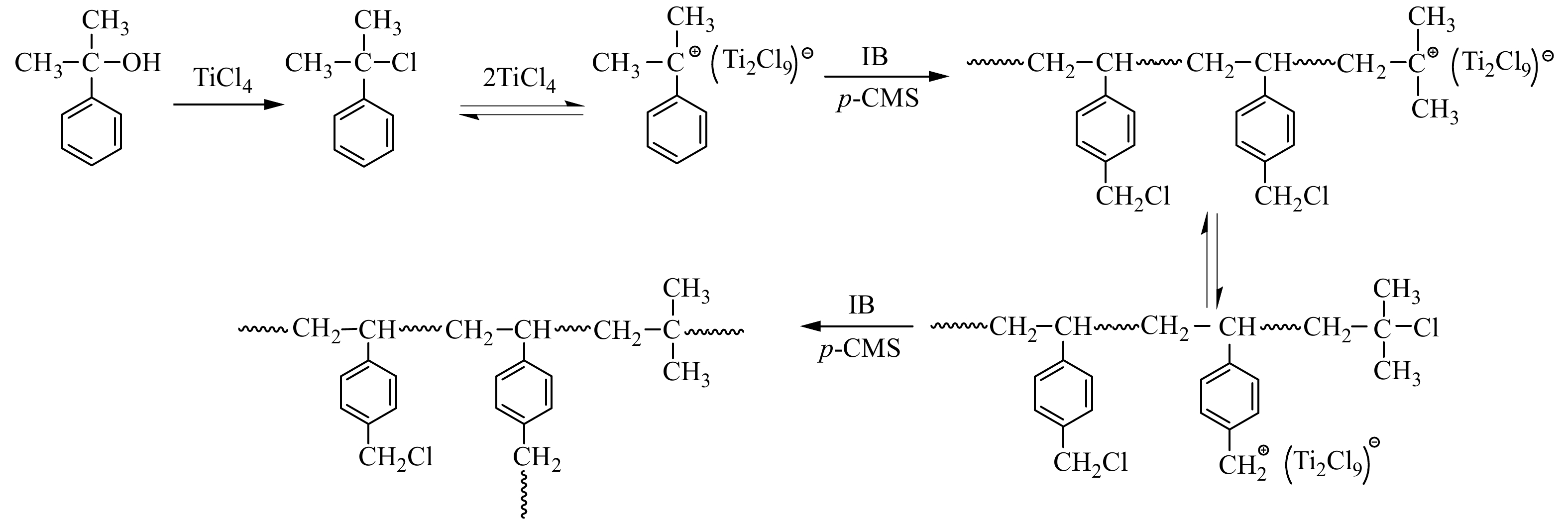
基于以上分析结果,初步归纳了TiCl4共引发p-CMS与IB正离子共聚合的机理(示意图1):TiCl4与枯基醇发生卤素交换,生成枯基氯,进而产生枯基碳正离子,引发共聚合;在聚合过程中,存在活性中心与叔丁基氯休眠种之间的平衡;随着共聚反应的进行,共聚到大分子链中的苄基氯缓慢参与引发,形成支化.
图 1
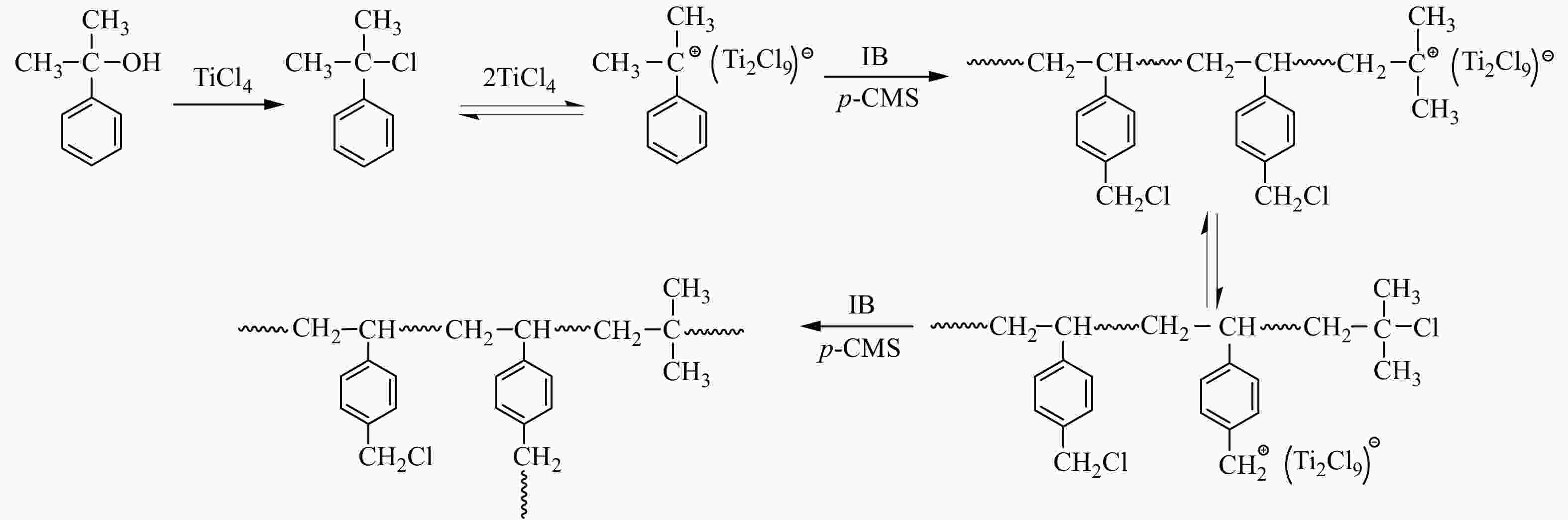 Figure 1. The proposed mechanism of IB and p-CMS copolymerization with CumOH/TiCl4 initiating system
Figure 1. The proposed mechanism of IB and p-CMS copolymerization with CumOH/TiCl4 initiating system3. 结论
在−80 °C下的n-Hex/CH2Cl2 (V/V = 6/4)的混合溶剂内,采用路易斯酸性相对较弱的TiCl4作为共引发剂时,可以得到无凝胶单峰分布的p-CMS-IB共聚物(CIMS). o-CMS不能参与共聚,p-CMS的共聚活性较低. 随反应时间延长和单体转化率的提高,共聚物中p-CMS的含量以及产物分子量均逐渐增加. 相比于高活性的枯基引发剂,p-CMS单体几乎不参与引发,但随反应进行,共聚到大分子链中的苄基氯缓慢参与引发,形成支化. 支化产物具有较低的玻璃化转变温度. 提高聚合温度,共聚合速率降低,但p-CMS的引发活性未发生明显变化. 众所周知,支化结构赋予聚合物特有的流变学和物理机械性能[33,34]. TiCl4共引发可合成出链中含有高活性苄基氯基团,且同时具有支化结构的CIMS,在链中官能化PIB以及主链全饱和气密层橡胶等领域具有研究和应用价值. 如何进一步提高共聚物分子量,以及对共聚物支化结构、流变学性能以及物理性能的详细研究正在进一步进行.
致谢 感谢北京化工大学吴一弦教授课题组友情提供的三检测GPC测试.
-
-
[1]
Malmberg S E, Parent J S, Pratt D A. Macromolecules, 2010, 20: 8456 − 8461
-
[2]
Xie Zhonglin(谢忠麟). Journal of Rubber Technology Market(橡胶科技市场), 2008, (11): 7 − 8
-
[3]
Jones G D. US patent, 3067182. 1962-11-04
-
[4]
Jones G D, Runyon J R, Ong J. J Appl Polym Sci, 1961, 16: 452 − 459
-
[5]
Jones G D, Runyon J R, Ong J. Ind Eng Chem, 1961, 53: 291 − 298
-
[6]
Powers K W. US patent, 4074035. 1978-03-14
-
[7]
Powers K W. US patent, 3948868. 1976-04-06
-
[8]
Wang H C. CN patent, CN1468282A. 2004-01-14
-
[9]
Wang H C. CN patent, CN1636036A. 2005-07-06
-
[10]
Nuyken O, Gruber F, Pask S D. Makromol Chem, 1993, 194: 3415 − 3432 doi: 10.1002/macp.1993.021941220
-
[11]
Nuyken O, Sanchez J R, Voit B. Macromol Rapid Commun, 1997, 18: 125 − 131 doi: 10.1002/marc.1997.030180209
-
[12]
Grasmuller M, Rueda-sanchez J C, Nuyken O. Macromol Symp, 1998, 127: 109 − 114 doi: 10.1002/masy.19981270115
-
[13]
Nuyken O, Vierle M. Des Monomer Polym, 2005, 8: 91 − 105 doi: 10.1163/1568555053603233
-
[14]
Osman A. US patent, 5629386. 1997-05-13
-
[15]
Schafer M, Wieland, P C, Nuyken O. J Polym Sci, Part A: Polym Chem, 2002, 40: 3725 − 3733 doi: 10.1002/pola.10472
-
[16]
Kennedy J P. US patent, 4327201. 1982-04-27
-
[17]
Yan P F, Guo A R, Liu Q, Wu Y X. J Polym Sci, Part A: Polym Chem, 2012, 50: 3383 − 3392 doi: 10.1002/pola.26126
-
[18]
Liu Q, Wu Y X, Yan P F, Zhang Y, Xu R W. Macromolecules, 2011, 44: 1866 − 1875 doi: 10.1021/ma1027017
-
[19]
Kanaoka S, Sawamoto M, Higashimura T. Macromolecules, 1996, 29: 1778 − 1783 doi: 10.1021/ma951320j
-
[20]
Kamigaito M, Nakashima J, Satoh K, Sawamoto M. Macromolecules, 2003, 36: 3540 − 3544 doi: 10.1021/ma0216876
-
[21]
Ren Ping(任苹), Wu yibo(伍一波), Guo wenLi(郭文莉), Li ShuXin(李树新), Wang HongLiang(王洪亮), Xiao Fei (肖菲), Liu KeLong(刘克龙). China Elastomerics(弹性体), 2012, 22(1): 25 − 28 doi: 10.3969/j.issn.1005-3174.2012.01.006
-
[22]
Kelen T, Tüdős F. J Macromol Sci Part A, 1975, A9(1): 1 − 27
-
[23]
Cao Lina(曹丽娜), Luo Qingzhi(罗青枝), Xiao Libin(肖丽彬), Wang Desong(王德松), Dou Haiyang(窦海洋). Journal of Hebei University of Science and Technology(河北科技大学学报), 2010, 31(3): 195 − 200 doi: 10.7535/hbkd.2010yx03003
-
[24]
Held D, Ivan B, Muller A H E. Macromolecules, 2001, 34: 2418 − 2426 doi: 10.1021/ma000641e
-
[25]
Ashbaugh J R, Ruff C R, Shaffer T D. J Polym Sci, Part A: Polym Chem, 2000, 38: 1680 − 1686 doi: 10.1002/(SICI)1099-0518(20000501)38:9<1680::AID-POLA34>3.0.CO;2-Y
-
[26]
Fodor Z, Bae Y C, Faust R. Macromolecules, 1998, 31: 4439 − 4446 doi: 10.1021/ma980193z
-
[27]
Fréchet J M J, Henmi M, Gitsov I, Ashima S, Leduc M R, Grubbs R B. Science, 1995, 269: 1080 − 1083 doi: 10.1126/science.269.5227.1080
-
[28]
Simon P F W, Muller A H E. Macromolecules, 2001, 34: 6206 − 6213 doi: 10.1021/ma002156p
-
[29]
Zhang C B, Zhou Y, Liu Q, Li S X, Perrier S, Zhao Y L. Macromolecules, 2011, 44: 2034 − 2049 doi: 10.1021/ma1024736
-
[30]
Wang Z M, He J P, Tao J F, Yang L, Jiang H J, Yang Y L. Macromolecules, 2003, 36: 7446 − 7452 doi: 10.1021/ma025673b
-
[31]
Hull D L, Kennedy J P. J Polym Sci, Part A: Polym Chem, 2001, 39: 1515 − 1524 doi: 10.1002/pola.1128
-
[32]
Safa K D, Babazadeh M. Eur Polym J, 2004, 40: 1659 − 1669 doi: 10.1016/j.eurpolymj.2004.04.005
-
[33]
Voit B I, Lederer A. Chem Rev, 2009, 109: 5924 − 5973 doi: 10.1021/cr900068q
-
[34]
Gao C, Yan D. Prog Polym Sci, 2004, 29: 183 − 275 doi: 10.1016/j.progpolymsci.2003.12.002
-
[1]
-
Figure 2 1H-NMR spectrum of the resulting copolymer (Conditions: CMS/IB = 2.5/100 (molar ratio). [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L, reaction time = 6 min, T = −80 °C)
Figure 4 Plots of conversion versus time for p-CMS-IB copolymerization (Conditions: [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L, T = −80 °C)
Figure 5 Plots of first-order plots of ln([M0]/[M]) versus time for p-CMS-IB copolymerization
Figure 11 Mark-Houwink-Sakurada plots for linear PIB and IB/p-CMS copolymer (run 0410-5 in Table 4) (LS data: Linear PIB, Mn = 3.9 × 104, Mw = 8.6 × 104, MWD = 2.19; Copolymer, Mn = 5.3 × 104, Mw = 12.5 × 104, MWD = 2.37)
Figure 1 The proposed mechanism of IB and p-CMS copolymerization with CumOH/TiCl4 initiating system
Table 1. The experimental phenomenon with different initiating systems
Initiator Co-initiator Electron donor Phenomenon CumOH TiCl4 − Polymer; no gel; pale yellow H2O AlEtCl2 − Polymer and gel; red H2O AlEt1.5Cl1.5 − Polymer and gel; red H2O AlEt2Cl − No polymer H2O AlCl3 Anisole No polymer H2O AlCl3 Chloroacetyl chloride Polymer and gel; red Reaction conditions: [co-initiator] = 0.045 mol/L, [CumOH] = 0.01 mol/L, ED/AlCl3 (molar ratio) = 1.1/1, n-Hex/CH2Cl2 = 6/4 (V/V), [IB] = 1.63 mol/L, [CMS] = 0.33 mol/L, T = − 80 °C, reaction time = 30 min  下载: 导出CSV
下载: 导出CSV
Table 2. Copolymer compositions by 1H-NMR
Run Conversion (%) Monomer feeding (%) Copolymer composition (%) IB p-CMS IB p-CMS − 100 0 100 0 1 11.4 97.88 2.12 99.54 0.46 2 9.5 91.65 8.35 97.96 2.04 3 13.9 82.95 17.05 95.36 4.64 4 8.5 73..95 26.05 90.94 9.06 5 7.56 64.60 35.40 88.13 11.87 6 7.3 54.89 45.11 82.02 17.98 − 0 100 0 100 Conditions: [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L, reaction time = 6 min, T = −80 °C
下载: 导出CSV
Table 3. sReactivity ratios of the p-CMS-IB copolymerization systems
Methods Positive sequence Reverse rIB rp-CMS rIB′ rp-CMS′ K-T 4.658 0.7195 4.6582 0.7196 YBR 4.676 ± 0.075671 0.6886 ± 0.02088 Average rIB = 4.67 rp-CMS = 0.70
下载: 导出CSV
Table 4. The 1H-NMR data of the resulting copolymers
Run Reaction time
(min)Mn,GPC,RI $F_{\rm CH_2 Cl} $
(mol%)ICMS
(mol%)Branching points
(per chain)BSB
(mol%)SSB
(mol%)0410-1 3 NA 0.69 0.69 0 100 0 0410-2 30 11700 0.8 1.08 0.58 100 0 0410-3 45 12680 0.82 1.23 0.93 72.46 27.53 0410-4 60 15670 0.76 1.45 1.93 70.92 29.08 0410-5 90 16220 0.89 2.13 3.59 59.17 40.83
下载: 导出CSV
Table 5. The monomer conversion and 1H-NMR data of the resulting copolymers
Run Reaction
time (min)Reaction
temp. (°C)Conversion
(%)$F_{{\rm CH}_2 {\rm Cl}} $
(mol%)ICMS
(mol%)BSB
(mol%)SSB
(mol%)1018-4 45 −40 9.62 NA NA NA NA 1018-6 90 −40 23.08 0.88 1.61 74.00 32.00 1018-8 20 −60 15.31 NA NA NA NA 1018-9 30 −60 20.19 0.66 0.99 59.49 40.51 1018-10 45 −60 26.12 NA NA NA NA 1018-11 60 −60 39.42 0.89 1.33 56.59 43.41 1018-12 90 −60 51.92 0.71 1.6 71.83 28.17 Conditions: CMS/IB = 5/100. [IB] = 1.63 mol/L, [CumOH] = 0.004 mol/L, [DTBP] = 0.005 mol/L, [TiCl4] = 0.045 mol/L
下载: 导出CSV
-














 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 102
- 文章访问数: 8423
- HTML全文浏览量: 1407
