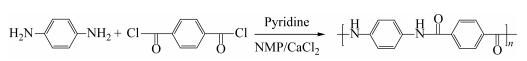
Scheme 1.
Polycondensation of PPD and TPC for the preparation of PPTA
Kinetics Analysis on the Polycondensation Process of Poly(p-phenylene terephthalamide): Experimental Verification and Molecular Simulation
Jing Liu , Hai-Juan Kong , Yu Ma , Shu Zhu , Mu-Huo Yu

Poly(p-phenylene terephthalamide) (PPTA) has been noted as a kind of important polymer materials which presents a combination of outstanding properties such as good chemical resistance, low flammability and excellent mechanical properties[1]. It also possesses high thermal stability with high melting temperature[2], and forms highly oriented structure because of their rigid backbones.
As an important polymer product with growing demand[1, 3-5], there have been extensive researches focusing on the PPTA synthesis in recent years[6-8]. The conventional preparation method for PPTA is low temperature solution polycondensation of p-phenylenediamine (PPD) and terephthaloyl chloride (TPC) in a mixed solvent of N-methyl-2-pyrrolidone (NMP) containing CaCl2, as shown in Scheme 1, which is the most widely used in almost all commercial products, like Kevlar, Twaron, Technora, etc[9]. CaCl2 is used to weaken the hydrogen bond interaction between PPTA chains because of the complexation between calcium ion and amidogen. In this cooling polycondensation process, it involves low initial reaction temperature and significant increase of viscosity with the molecular weight growing, resulting in severe difficulty in reactant mixing, also leading to strict temperature control demand and high blending requirements[10]. Because of the high-activity monomers, this process requires strict dehydration, deoxidation and accurate temperature control. Pyridine is necessarily used for the absorption of HCl because of its alkaline effect, avoiding the side reaction between HCl and PPD. Moreover, the polycondensation will be completed quickly because the activation energy was is low (6.3 kJ/mol). Due to the strongly exothermic nature (reaction enthalpy is -199 kJ/mol)[11], this process usually results in a rapid increase in the temperature.
PPTA is commonly transformed into high-performance fibers and fabrics to be used as transformed materials and composite materials with superior thermal and mechanical resistance. There is still wide research interest in obtaining PPTA with high molecular weight to be transformed into high tenacity and modulus fibers. However, only about 15% of the theoretical tensile strength has been achieved, while fibers with tensile modulus up to 50%-80% of the theoretical value have been commercialized[12]. The tensile strength is highly dependent on defects including the chain ends, whereas the tensile modulus is not very sensitive to this. The molecular weight and the obtained chain length have been shown to be directly related to the tensile strength of high modulus polymeric fibers. However, molecular weight cannot be increased without limit by using current technologies because of difficulties in polymerization and processing.
The polycondensation of PPTA follows the step-reaction mechanism that any two oligomers with the appropriate functional groups can react to form a larger molecule[13], and become strong diffusion-controlled at the moderate degree of polymerization[14]. This diffusion effect retards the primary reaction and prevents the formation of high molecular weight polymer due to the capping of functional groups by side reactions that become significant. However, this diffusion-effected kinetics of polycondensation of PPTA is difficult to be detected by experiment because of the fast reaction rate and the orientation constraint of rod-like polymers. A number of previous works have been carried out to study the polymerization kinetics of rod-like molecules under quiescent conditions or well defined flow conditions by means of experiments and theory[15-23] based on the Smoluchowski' diffusion equation[24]. It described the polymerization kinetics by using three kinds of diffusion coefficient about the axial direction, radial direction and rotational. Many theories for the rotational and translation diffusion coefficients for the rigid rod-like polymers, like PPTA, in dilute solution modeled the molecule as a smooth cylinder, thus the complex geometry of the molecular surface could be ignored[25]. These theoretical approaches also assumed "stick" (nonslip) boundary conditions at the surface of the particle. In the semi-dilute solution regime, the theoretical studies were based on the "tube model" proposed by Doi[26] and the general expression obtained by Teraoka that could be applied in all over the concentration range[27, 28]. Agarwa and Khakhar[15-17] developed the above theories and studied the polymerization and dynamics of rigid rod-like polymers both in dilution and semi-dilution solution, especially about the condensation polymerization of PPTA. The obtained relationship between reaction rate constant, monomer concentration, degree of polymerization and shearing rate established the foundation for the further study of kinetics research of PPTA. Agge and Khakhar[22, 29, 30] studied the kinetics of solution polymerization to synthesize PPTA theoretically and experimentally under quiescent conditions or extensional flow conditions. These studies presented an integrated kinetics of solution polymerization of PPTA. However, these research works only revealed the polymerization rate constant of polymers with the same chain length, rather than the polydispersed system.
In this paper, the conventional preparation method for PPTA was described in detail. Since PPTA with higher molecular weight will build up better mechanical properties and thermostability so that can be applied to different application fields, the molecular weight is the most important criterion for PPTA. The effects of different experimental conditions on the inherent viscosity (ηinh) and the number-average degree of polymerization (
p-Phenylenediamine (PPD, purity > 99.9%) and terephthaloyl chloride (TPC, purity > 99.9%) were purchased from J & K Chemical Co. Ltd. N-methyl-2-pyrrolidone (NMP, purity > 99.9%), calcium chloride anhydrous (CaCl2, purity > 96%), pyridine (Py, purity > 99.9%) and sulfuric acid (H2SO4, purity > 98%) were all purchased from Sinopharm Chemical Reagent Co. Ltd, China. The trace water in NMP was removed by adding NMP in molecular sieve and CaH2 before the experiments. CaCl2 was carefully heated for 4 h at 400 ℃ to dehydrate in a muffle furnace before use. The oxygen content of the nitrogen used in the process is less than 3 ppm.
The temperature was controlled by a refrigerated circulating bath, which was bought from Shanghai Bilon Instrument Company. An Ubbelohde viscometer with capillary diameter 0.9-1.0 mm was used to measure the inherent viscosity of PPTA at 30 ℃.
The polymerization was carried out in a jacketed glass reactor. Initially, 500 mL of dried NMP was added into the 2 L reaction vessel, which had been purged by nitrogen to remove oxygen. The reaction system was heated to 78 ℃. Then, finely ground dry CaCl2 was added into the NMP solution with stirring. After CaCl2 was completely dissolved, PPD and a certain amount of pyridine were added to the NMP/CaCl2 solution with stirring. When all PPD and pyridine were completely dissolved, an ice-water bath was used to absorb heat and cool the solution to -10~5 ℃. The polymerization was initiated at this stage by adding powdered TPC under rapid stirring. The TPC was divided into two equal parts and added in sequence to react with PPD with a 10 min interval. The whole reaction process was carried out under a nitrogen atmosphere. The precipitated PPTA was placed in the grinder, and washed with deionized water at high-speed for several times so that the reaction was quenched. Then the polymer was immersed in ethanol for 8 h to remove water and NMP absorbed in PPTA. Finally, the samples were dried overnight under vacuum at 80 ℃ before being tested. The yield of PPTA was higher than 90%.
The molecular weight was measured by inherent viscosity method. The obtained PPTA (0.25 g) was initially dissolved in 98% sulfuric acid at 70 ℃ to prepare a PPTA/H2SO4 solution with a concentration of 0.5 g/dL and then measured at (30 ± 0.1) ℃ using an Ubbelohde viscometer. By calculating the ratio of the flow times between solvent and solution, the relative viscosity and consequently the inherent viscosity were determined as a measure for the molecular weight of PPTA. The inherent viscosity was obtained as
|
${\eta _{{\rm{inh}}}} = \frac{1}{c}\ln \left( {{\eta _{\rm{r}}}} \right) = \frac{1}{c}\ln \left( {\frac{t}{{{t_0}}}} \right)$ |
where ηinh is the inherent viscosity; ηr is the relative viscosity; t represents the efflux time for the polymer solution (PPTA/H2SO4), t0 represents the efflux time for the 98% sulfuric acid, and c represented the mass concentration of the PPTA/H2SO4 solution (g/dL).
The relationship between inherent viscosity and weight-average molecular weight of PPTA is[31]
|
${M_{\rm{w}}} = 3165\eta _{{\rm{inh}}}^{{\rm{1}}{\rm{.503}}}$ |
where ηinh ranges from 2.6 dL/g to 9.0 dL/g.
The weight-average degree of polymerization was calculated by
|
${\overline {{\rm{DP}}} _{\rm{w}}} = {M_{\rm{w}}}/238$ |
where 238 is the molecular weight of repeat unit of PPTA.
According to the relationship between the number-average degree of polymerization (
|
${\bar X_{\rm{n}}} = \frac{1}{{1 - p}}$ |
|
${\overline {{\rm{DP}}} _{\rm{w}}} = \frac{{1 + p}}{{1 - p}}$ |
The number-average degree of polymerization was calculated by
|
${\bar X_{\rm{n}}} = \frac{{{{\overline {{\rm{DP}}} }_{\rm{w}}} + 1}}{2}$ |
X-ray diffraction (XRD) was used to determine the crystal unit cell parameters and the apparent lateral crystal size. The XRD measurements were carried out on a Rigaku X-ray diffractometer (D/MAX-2550PC, Rigaku, Tokyo, Japan). These PPTA samples prepared to be powder were laid on the glass sample holder (35 mm × 50 mm × 5 mm). Ni-filtered Cu Kα radiation (λ = 0.154 nm) generated at a voltage of 40 kV and current of 35 mA was utilized at a scan speed of 5 (°)/min from 5° to 90°. The step-scan method was used to determine the d spacing and stacking size (D). The step-interval was set at 0.02°. The data were analyzed with Peakfit software (version 4.12, Seasolve Co., San Jose, CA). The d space and D were calculated by using Eq. (7) (the Bragg equation) and Eq. (8) (the Scherrer equation), respectively:
|
$n\lambda = 2d{\rm{sin}}\theta $ |
|
$D = K\lambda /B{\rm{cos}}\theta $ |
in which λ is X-ray wavelength (λ = 0.154 nm), K is the Scherrer constant (= 1.0), and B is the full-width at half-maximum of the reflection measured in 2θ, where θ is the corresponding Bragg angle. We neglected corrections for instrumental broadening in the calculation.
The crystallinity was calculated from the areas of crystalline diffraction peaks and the amorphous area using Hinrichen's method as[32]:
|
${C_{\rm{r}}} = {A_{\rm{c}}}/({A_{\rm{a}}} + {A_{\rm{c}}})$ |
where Ac is the area of crystalline peaks and Aa is the area of amorphous peak.
We performed dynamic Monte Carlo simulation to study the polymerization of PPTA. The 26214, 78644, 131072, 183502, 235930 monomers were preset in a 64 × 64 × 64 lattice box to reach the occupation density 0.1, 0.3, 0.5, 0.7, 0.9, respectively according to our previous study[33]. Generally, each monomer occupied a single lattice site, and a chain like polymer could be mimicked by consecutive occupied sites on the cubic lattice linked with bonds. All monomers were predefined as bifunctional, containing two reactive end groups, which could form bonds by reacting with each other. Monomers underwent athermal relaxation over a long enough period to become the bulk amorphous phase, and then adopted the sampling algorithm to determine whether a new conformation could be accepted.
In our algorithm, new configuration was obtained by both monomer movements and reactions. The movement was accomplished by the model with single-site jumping in company with periodic boundary conditions. Chain dynamics followed the relaxation algorithm that was in good accordance with both the Rouse model in dilute solutions and the reptation models in concentrated one. Double monomer occupation and bond crossing were forbidden to mimic the volume exclusion of polymer chains.
For the reaction part, a monomer and a random neighbor site of it were chosen to check whether these two sites could be linked with a new bond. It must be noted that the selected two monomers should contain opposite reactive groups and the reaction within one molecule was forbidden. In the algorithm, the reaction probability kMC could be employed to study the kinetics of polymerization. It is defined as the acceptance ration of reaction and directly analogous to
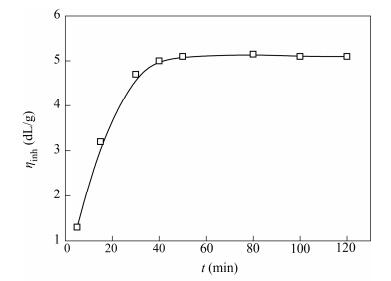
The monomer concentration had a great influence on ηinh of PPTA. As shown in Fig. 1, the range from 0.2 mol/L to 0.6 mol/L of monomer concentration was investigated to study the effect of monomer concentration on PPTA. The range from 0.35 mol/L to 0.45 mol/L was a good choice for the preparation of PPTA with higher molecular weight. When the monomer contraction was low, some side reactions, like the hydrolysis of TPC with trace amounts of water and oxidation of PPD with trace amounts of oxygen, would prevent the growth of molecular chains. However, when the monomer concentration was too high, it would cause an ultra-fast reaction rate so that the solubility of polymers decreased to form the gel, causing the reactive groups to be embedded in them and difficult to diffuse, which would be adverse to further polymer chain growth. It needs large energy input to mix the gelatinous or solid-state polymers. Furthermore, the ultra-fast reaction rate, caused by high monomer concentration, could also generate immense heat that gave rise to side reactions, such as branching and crosslinking. When the monomer concentration was proper, there would be an appropriate collision probability between monomers, oligomers and polymers, which was also beneficial to obtaining PPTA with high molecular weight.
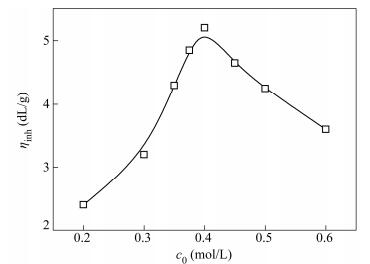
The effect of reaction temperature on ηinh of PPTA is shown in Fig. 2. It can be seen that the number-average degree of polymerization increased with the increase of reaction temperature, but decreased when the temperature ranged from -5 ℃ to 15 ℃. It can also be found that the molecular weight decreased when the temperature ranged from 0 ℃ to 15 ℃. This behavior was because the polycondensation of TPC and PPD, which had low activation energy and high reaction rate constant (1000 L/(mol·s)), would generate a lot of heat (about 199 kJ)[35] and make the reaction temperature rapidly increase about 20 ℃ in 10 s at the initial stage of reaction[15]. The high reaction temperature and intense reaction heat would lead to premature precipitation of PPTA gel, which was unfavorable for the preparation of PPTA with high molecular weight. In addition, high reaction temperature could also lead to oxidation of PPD and the side reactions of TPC, which would not only destroy monomer molar ratio, but also increase the risk of adverse reactions, and lead to the formation of by-products. If the reaction temperature was too low, it would cause the imbalance of the reaction ratio due to the decreased solubility of monomers in solvent and also prevent obtaining high molecular weight polymers owing to the low reaction rate. As a result, the low temperature ranging from -5 ℃ to 5 ℃ was the best condition to achieve PPTA with high molecular weight.
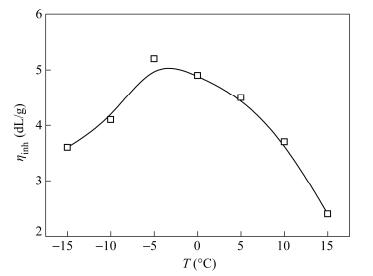
Figure 3 presents the effect of reaction time on ηinh and
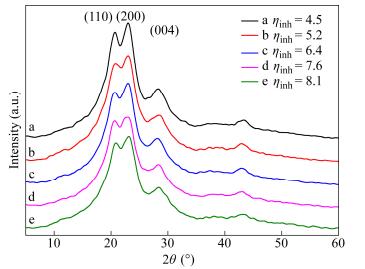
Based on the optimal conditions as mentioned above, PPTA samples with different inherent viscosities were obtained by the twin screw. The detailed technological parameters for each reaction step within the twin screw will be discussed in another article. In the present study, microstructure parameters of the prepared PPTA resin were investigated by X-ray diffraction technique. An X-ray beam directed perpendicularly to the axis produced reflections from layer planes. The Scherrer equation was used to calculate the stacking height of layer planes in vertical direction of the crystal. Figure 4 presents equatorial X-ray diffraction patterns of PPTA with different inherent viscosities recorded at room temperature. Qualitative inspection of equatorial X-ray diffraction patterns showed three sharp peaks at about 2θ =20.9° (d = 0.42 nm), 2θ = 23.6° (d = 0.39 nm) and 2θ = 28.5° (d = 0.31 nm), which could be indexed as (110), (200) and (004) diffractions, respectively[36, 37]. Crystal parameters obtained from equatorial X-ray diffraction patterns are given in Table 1. It indicates that the crystallinity increased with the inherent viscosity but the stacking size (D) of PPTA was almost invariable even with different inherent viscosities and crystallinities.
 DownLoad:
CSV
DownLoad:
CSV
| ηinh (dL/g) |
Crystallinity (%) |
(110) | (200) | (004) | |||||
| 2q (°) | D (Å) | 2q (°) | D (Å) | 2q (°) | D (Å) | ||||
| 4.5 | 48.69 | 20.6 | 32 | 23.0 | 30 | 28.3 | 23 | ||
| 5.2 | 51.53 | 20.7 | 28 | 23.1 | 29 | 28.5 | 20 | ||
| 6.4 | 54.53 | 20.6 | 29 | 23.0 | 31 | 28.2 | 23 | ||
| 7.6 | 57.40 | 20.6 | 31 | 22.9 | 27 | 28.4 | 24 | ||
| 8.1 | 59.75 | 20.9 | 30 | 23.2 | 26 | 28.4 | 21 | ||
To better understand the kinetics of polycondensation for preparing PPTA, a kinetics model in this work was built based on our previous work[33]. In this model, the reaction probability kMC, which is defined as the acceptable ration of reaction, was employed to study the kinetics of polymerization. It would be proportional to the apparent reaction rate kapp, and could be used to control the rate of reaction.
In the early stage of polycondensation, viscosity of the reaction system was not very high which assured the free movement of molecules in the system. The reaction between amine groups and acyl chloride groups strictly obeyed the second order reaction kinetics, where the molecular weight increased linearly with the progress of polymerization. However, when the reaction probability increased, the number-average degree of polymerization (
|
${\bar X_{\rm{n}}} \sim c_0^{ - 0.88} \cdot {t^{0.37}}$ |
Thus the number-average degree of polymerization (
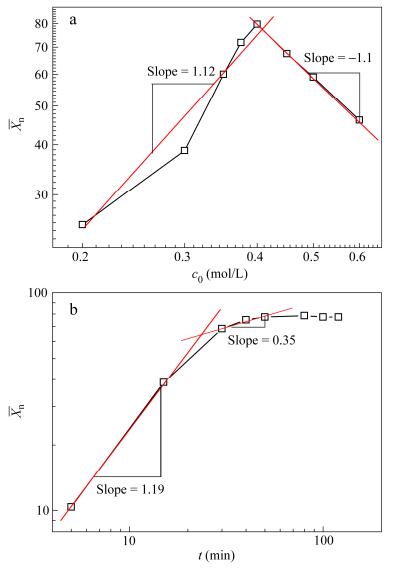
As shown in Fig. 5(a), the solution polymerization was investigated from the
With the increasing monomer concentration, a diffusion constant strongly dependent on chain length altered this situation. The chain lengths increased longer in higher concentration so that their rotational diffusion constants asymptotically approached zero. The time required to form the transition state necessary for condensation was longer than the lifetime for reaction. The rate-limiting step would be the formation of collinear alignment transition state. In the extreme, all molecules achieved co-linearity and proximity condense by diffusion and the polymerization rate was controlled by the rate at which this transition state was formed. Furthermore, a similarly definitive analysis about reaction or diffusion by reaction rate theory of relating the reaction probability to the time has been given in our previous study. It could be found that the scaling of concentration rapidly decreased because the entanglements restricted the diffusion within a primitive tube. The scaling exponent between
With the double logarithmic plots of
Further examination of the polycondensation process was carried out with the
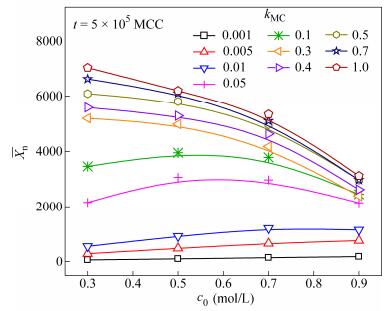
In this work, the polycondensation reaction between p-phenylenediamine (PPD) and terephthaloyl chloride (TPC) was performed to synthesize poly(p-phenylene terephthalamide) (PPTA). Effects of a number of factors, including monomer concentration, reaction temperature and reaction time on the molecular weight of PPTA were investigated to determine the optimum conditions for preparing high molecular weight PPTA. Furthermore, we studied the kinetics of polycondensation with theoretical analysis and dynamic Monte Carlo simulation. The theoretical results for the variation of the degree of polymerization with time showed a typical behavior of a diffusion-controlled stepwise polymerization with severe slowing of the reaction at the later stages. The simulation result showed the number-average degree of polymerization to be of the form
Rao Y., Waddon A., Farris R.. Structure-property relation in poly(p-phenylene terephthalamide) (PPTA) fibers[J]. Polymer, 2001, 42(13): 5937-5946. doi: 10.1016/S0032-3861(00)00905-8
Mark H., Atlas S., Ogata N.. Aromatic polyamide[J]. J. Polym. Sci., 1962, 61(172): 49-53. doi: 10.1002/pol.1962.1206117225
Anagnostopoulos G., Parthenios J., Galiotis C.. Thermal stress development in fibrous composites[J]. Mater. Lett., 2008, 62(3): 341-345. doi: 10.1016/j.matlet.2007.05.031
Knijnenberg A., Bos J., Dingemans T. J.. The synthesis and characterisation of reactive poly(p-phenylene terephthalamide)s:a route towards compression stable aramid fibres[J]. Polymer, 2010, 51(9): 1887-1897. doi: 10.1016/j.polymer.2010.03.015
Rao Y., Waddon A., Farris R.. The evolution of structure and properties in poly(p-phenylene terephthalamide) fibers[J]. Polymer, 2001, 42(13): 5925-5935. doi: 10.1016/S0032-3861(00)00906-X
Du S., Wang W., Yan Y., Zhang J., Tian M., Zhang L., Wan X.. A facile synthetic route to poly(p-phenylene terephthalamide) with dual functional groups[J]. Chem. Commun., 2014, 50(69): 9929-9931. doi: 10.1039/C4CC02366H
Du S., Zhang J., Guan Y., Wan X.. Sequence effects on properties of the poly(p-phenylene terephthalamide)-based macroinitiators and their comb-like copolymers grafted by polystyrene side chains[J]. Aust. J. Chem., 2014, 67(1): 39-48. doi: 10.1071/CH13291
Schwartz P.. A review of recent experimental results concerning the strength and time dependent behavior of fibrous poly(paraphenylene terephthalamide)[J]. Polym. Eng. Sci., 1987, 27(11): 842-847. doi: 10.1002/(ISSN)1548-2634
Perepelkin K. E., Machalaba N. N.. Recent achievements in structure ordering and control of properties of para-aramide fibres[J]. Mol. Cryst. Liq. Cryst., 2000, 353(1): 275-286. doi: 10.1080/10587250008025667
Sun L., Xu J., Luo W., Guo C., Tuo X., Wang X.. Investigation on the preparation of high molecular weight poly(p-phenylene terephthalamide) using CaH2 as acid absorbent[J]. Acta Polymerica Sinica (in Chinese), 2012, (1): 70-74.
Wang S., Liu H., Xiao R.. Determination of condensation-polymerization thermal effect of poly(paraphenylenetere-phalamide)[J]. Journal of DongHua University (in Chinese)., 1984, 1: 41-46.
Chae H. G., Kumar S.. Rigid-rod polymeric fibers[J]. J. Appl. Polym. Sci., 2006, 100(1): 791-802. doi: 10.1002/(ISSN)1097-4628
Flory, P. J., Principles of polymer chemistry, Cornell University Press, New York, 1953, p. 317.
Cotts D. B., Berry G. C.. Polymerization kinetics of rigid rodlike molecules:polycondensation of poly([benzo (1, 2-d:5, 4-d') bisoxazole-2, 6-diyl]-1, 4-phenylene)[J]. Macromolecyles, 1981, 14(4): 930-934. doi: 10.1021/ma50005a008
Agarwal U., Khakhar D.. Enhancement of polymerization rates for rigid rod-like molecules by shearing[J]. Nature, 1992, 360: 53-55. doi: 10.1038/360053a0
Agarwal U., Khakhar D.. Diffusion-limited polymerization of rigid rodlike molecules:dilute solutions[J]. J. Chem. Phys., 1992, 96(9): 7125-7134. doi: 10.1063/1.462546
Agarwal U., Khakhar D.. Shear flow induced orientation development during homogeneous solution polymerization of rigid rodlike molecules[J]. Macromolecules, 1993, 26(15): 3960-3965. doi: 10.1021/ma00067a035
Agarwal U., Khakhar D.. Simulation of diffusion-limited step-growth polymerization in 2D:effect of shear flow and chain rigidity[J]. J. Chem. Phys., 1993, 99(4): 3067-3074. doi: 10.1063/1.466195
Agarwal U., Khakhar D.. Diffusion-limited polymerization of rigid rodlike molecules:semidilute solutions[J]. J. Chem. Phys., 1993, 99(2): 1382-1392. doi: 10.1063/1.465382
Arpin M., Strazielle C.. Characterization and conformation of aromatic polyamides:poly(1, 4-phenylene terephthalamide) and poly(p-benzamide) in sulphuric acid[J]. Polymer, 1977, 18(6): 591-598. doi: 10.1016/0032-3861(77)90061-1
Bair T., Morgan P., Killian F.. Poly(1, 4-phenylenetere-phthalamides). polymerization and novel liquid-crystalline solutions[J]. Macromolecules, 1977, 10(6): 1396-1400. doi: 10.1021/ma60060a042
Gupta J. S., Agge A., Khakhar D.. Polymerization kinetics of rodlike molecules under quiescent conditions[J]. AlChE J., 2001, 47(1): 177-186. doi: 10.1002/(ISSN)1547-5905
Bao J. S., You A. J., Zhang S. Q., Zhang S. A., Hu C.. Studies on the semirigid chain polyamide-poly(1, 4-phenylenetere-phthalamide)[J]. J. Appl. Polym. Sci., 1981, 26(4): 1211-1220. doi: 10.1002/app.1981.070260413
Doi, M. ; Edwards, S. F., The theory of polymer dynamics, Oxford University Press, New York, 1988, p. 295.
Tracy M., Pecora R.. Dynamics of rigid and semirigid rodlike polymers[J]. Annu. Rev. Phys. Chem., 1992, 43(1): 525-557. doi: 10.1146/annurev.pc.43.100192.002521
Doi M.. Molecular dynamics and rheological properties of concentrated solutions of rodlike polymers in isotropic and liquid crystalline phases[J]. J. Polym. Sci., Part B, 1981, 19: 229-243.
Teraoka I., Hayakawa R.. Theory of dynamics of entangled rod-like polymers by use of a mean-field green function formulation.Ⅰ. transverse diffusion[J]. J. Chem. Phys., 1988, 89(11): 6989-6995. doi: 10.1063/1.455325
Teraoka I., Hayakawa R.. Theory of dynamics of entangled rod-like polymers by use of a mean-field green function formulation.Ⅱ. rotational diffusion[J]. J. Chem. Phys., 1989, 91(4): 2643-2648. doi: 10.1063/1.456973
Agge A., Jain S., Khakhar D.. Acceleration of the polymerization of rodlike molecules by flow[J]. J. Am. Chem. Soc., 2000, 122(44): 10910-10913. doi: 10.1021/ja001541r
Jain S., Agge A., Khakhar D.. Flow enhanced diffusion-limited polymerization of rodlike molecules[J]. J. Chem. Phys., 2001, 114(1): 553-560. doi: 10.1063/1.1330211
Zhang R., Kong H. J., Zhong H. P., Liu J., Zhou J. J., Teng C. Q., Ma Y., Yu M. H.. N-Alkyl PPTA:preparation and characterization[J]. Adv. Mater. Res., 2012, 554: 105-109.
Fitzer E., Müller D.. The influence of oxygen on the chemical reactions during stabilization of pan as carbon fiber precursor[J]. Carbon, 1975, 13(1): 63-69. doi: 10.1016/0008-6223(75)90259-6
Liu J., Ma Y., Wu R., Yu M.. Molecular simulation of diffusion-controlled kinetics in stepwise polymerization[J]. Polymer, 2016, 97: 335-345. doi: 10.1016/j.polymer.2016.05.050
Atkins, P. ; Paula, D. J. Physical Chemistry, W. H. Freeman & Company, New York, 2006, p. 807.
Wang S., Liu H., Xiao R.. Determination of condensation-polymerization thermal effect of poly(paraphenylenetere-phalamide)[J]. Journal of DongHua. University, 1984, 1: 41-46.
Northolt M.. X-ray diffraction study of poly(p-phenylene terephthalamide) fibres[J]. Eur. Polym. J., 1974, 10(9): 799-804. doi: 10.1016/0014-3057(74)90131-1
Northolt M., van Aartsen J.. On the crystal and molecular structure of poly-(p-phenylene terephthalamide)[J]. J. Polym. Sci., Part C:Polym. Lett., 1973, 11(5): 333-337. doi: 10.1002/pol.1973.130110508
Bu Z., Russo P. S., Tipton D. L., Negulescu I. I.. Self-diffusion of rodlike polymers in isotropic solutions[J]. Macromolecules, 1994, 27(23): 6871-6882. doi: 10.1021/ma00101a027
Wang P., Wang K., Zhang J.. Non-aqueous suspension polycondensation in NMP-CaCl2/paraffin system-A new approach for the preparation of poly(p-phenylene terephthalamide)[J]. Chinese J. Polym. Sci., 2015, 33(4): 564-575. doi: 10.1007/s10118-015-1607-1

 扫一扫看文章
扫一扫看文章

扫一扫关注我们