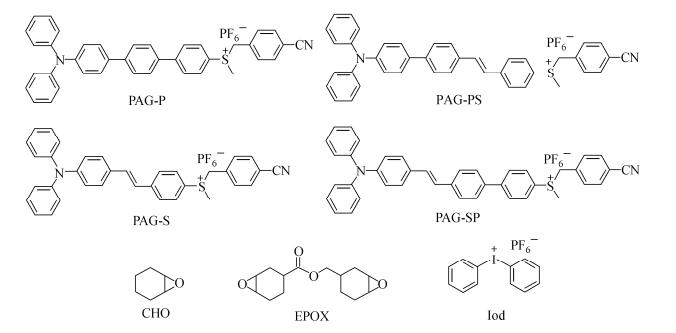
 Figure Scheme 1.
Molecular structures of PAGs, monomers, and iodonium salt
Figure Scheme 1.
Molecular structures of PAGs, monomers, and iodonium salt
Citation:
Xing-yu Wu, Jin Ming, Jian-chao Xie, De-cheng Wan, Malval Jean Pierre. Effects of Conjugated Systems on UV-Visible Light-sensitive D-π-A Type Sulfonium Salt Photoacid Generators[J]. Chinese Journal of Polymer Science,
2016, 34(12): 1456-1468.
doi:
10.1007/s10118-016-1863-8

Effects of Conjugated Systems on UV-Visible Light-sensitive D-π-A Type Sulfonium Salt Photoacid Generators
English
Effects of Conjugated Systems on UV-Visible Light-sensitive D-π-A Type Sulfonium Salt Photoacid Generators
Abstract:
A series of D-π-A type sulfonium salt photoacid generators with different π-conjugated structures, such as triphenyl, phenylstilbene, styryl-biphenyl, and stilbene, were designed to determine the effect of molecular structures on the photochemical and photophysical properties. The mechanisms of photochemical generation of H+ were studied by UV-Vis spectroscopy, theoretical calculations, and fluorescence spectroscopy. It is found that the frontier orbits determine the absorption, the molar extinction coefficients, and the quantum yields of photoacid generation. Triphenyl systems connected with sulfonium are beneficial to increase the quantum yields of acid generation. The photoreactivity of four sulfonium salts was further evaluated through the polymerizations of various epoxide monomers at different irradiation wavelengths (365-425 nm) by using the real-time infrared spectroscopy with light-emitting diodes. The high quantum yields for acid generation (ΦH+=~0.32 to 0.58) and the high molar extinction coefficients (ε=~23500 L·mol-1·cm-1 to 31000 L·mol-1·cm-1) of the sulfonium salts lead to high conversion rates (over 50%-80%). Hence, these photoinitiators exhibit potential for the photocuring applications.
-
Key words:
- Sulfonium salt
- / Photoacid generators
- / π-conjugated structures
-
INTRODUCTION
The photochemistry of onium salts, such as commonly used photoacid generators (PAGs) of iodonium[1] and sulfonium[2, 3], have been well established[4] and applied in printing, coatings, adhesives, microelectronics, stereolithography, and holography[5]. The discovery of highly photosensitive (i.e., high quantum yield) and useful (i.e., cationic photoinitiators) PAGs is crucial to the development of photochemistry[6]. Generally, commercial salts exhibit poor spectral sensitivity at long wavelength ( > 320 nm), thereby limiting their applications, particularly in the pigmented systems. Based on the technological advances of light-emitting diodes (LEDs)[7], onium salts are adapted to operate under LED irradiation, especially in the near UV/visible region[8-12]. These eco-friendly sources exhibit several advantages, including low cost, lack of harmful UV rays, and high operating efficiency[13].
Wavelength tunability has been partially solved by the photosensitive methods. Depending on the electron transfer-induced photosensitization, the spectral response of bimolecular systems can be extended into the near-UV and visible spectral regions[14-16]. However, the global kinetics of this bimolecular method is intrinsically limited by diffusion. As such, an integrated approach has been alternatively proposed[17-23]. Saeva[17], Marder[18], and Belfield[19] et al. reported several sulfonium salt-based PAGs with large π-conjugated systems in the structures. The bond photolysis and photoacid generation rates of sulfonium salts developed by Saeva[3, 24] and Marder[25] are dependent on three factors: (1) the energy of the excited state, (2) the stability of the radical cation, and (3) the stability of the neutral radical. The production of sufficient energy to photolyze sulfonium salts is the first step. The radical cation and neutral radical can be stabilized by electron resonance in the systems. For example, triphenylamino-containing radical cation can transfer the charge to nitrogen atom for stabilizing the radical cation, which is immediately formed upon homolysis of the sulfur-carbon (S-C) bond of the aryl dialkkylsufoium salt[3]. The 4-cyanobenzyl radical exhibits higher leaving propensity than benzyl and methyl because of its stability[20].
In this paper, we attempt to modify the π-conjugated systems of the parent phenylsulfonium salt structure by using triphenylamino substituent as an electron-pushing group and 4-cyanobenzyl-containing sulfonium as the electron-withdrawing groups (Scheme 1). Based on the photochemical and photophysical properties of PAGs, the influence of the π-conjugated systems on the photolysis and photoacid generation of these molecules was elucidated. The applications of PAGs as the photoinitiators in the cationic photopolymerization were also discussed.
Figure Scheme 1.
Molecular structures of PAGs, monomers, and iodonium salt
EXPERIMENTAL
Synthesis
The synthetic route is shown in Schemes 2 and 3. Intermediate products, namely, 4-bromophenyl 4-cyanobenzyl sulfane (1)[28], 4-(N, N-diphenylamino)-4′-bromobiphenyl (2)[23], 4-(4-(N, N′-diphenylamino) phenyl) phenylboronic acid (3)[23], diethyl 4-bromobenzylphosphonate (6)[25], 4-bromo-4′-(N, N′-diphenylamine) stilbene (7)[29], and 4-(N, N′-diphenylamino) styrene (9)[30], were prepared according to the procedures reported in the literature. In the Suzuki and Heck reactions, precursors were reacted with CF3SO3CH3 to obtain sulfonium salts, with CF3SO3- as anions. To study these PAGs as the cationic photoinitiators, we replaced the initial counterion CF3SO3- with PF6- according to an ion-exchanging method described in the literature[25]. Zhou et al.[25] demonstrated that the cationic photopolymerization of epoxide resins led to an efficient epoxy ring-opening reaction, with PF6-, not CF3SO3-, as the counterion.
General Instrumentations
Proton and carbon nuclear magnetic resonance spectra (1H-, 13C-NMR) were recorded on a BrukerAvance 500 (500 MHz) spectrometer. Chemical shifts were reported in parts per million (δ) downfield from the Me4Si resonance, which was used as the internal standard when recording NMR spectra. Mass spectra were obtained on a Micromass GCTTM.
UV-Vis spectra were recorded on a Mapada UV-6300 spectrophotometer. The photodecomposition process in a solution of acetonitrile were monitored by the UV-Vis spectra under a LED point curing (Uvata, Shanghai) irradiation of 365 nm (0.4 mW×cm−2). Steady-state fluorescence spectra were collected from a Hitachi-2700 spectrofluorometer. Emission spectra were spectrally corrected, and fluorescence quantum yields include the correction because the solvent refractive index were determined relative to quinine bisulfate in 0.05 mol×L-1 sulfuric acid (Фfl=0.52)[20].
Quantum yields for acid generation were measured under LED irradiation at 365 nm. All the irradiated PAGs dissolved in acetonitrile were previously N2 degassed. The progress of the photoreaction was monitored via UV-Vis absorption spectroscopy. The absorbance at the excitation wavelength must be higher than 2.5 before assuming a total absorption of the incident photons. The dose rates were maintained sufficiently small such that the changes in absorbance were lower than 10%. Rhodamine B (RhB) was used as a sensor of photoacid generation[26]. Acid generation in acetonitrile was also evaluated from the calibration curve of RhB, which was gradually protonated by addition of π-toluenesulfonic acid. Incident light intensity was measured through ferrioxalateactinometry.
Molecular orbital calculations were performed using the GAUSSIAN 09 package[27]. The electronic absorption spectra for different compounds were determined by time-dependent density functional theory (TD-DFT) at B3LYP/6-31G*. Molecular orbits involved in these transitions were then extracted, and the geometries were frequency checked.
The kinetics of the photopolymerization was monitored in situ by Fourier transform real-time infrared spectroscopy (FT-RTIR) with a Thermo-Nicolet iS5 instrument infrared spectrometer. The photosensitive formulations were deposited on a KBr pellet (40 μm thick) for irradiation at different wavelength LEDs. The evolution of the epoxy group content in the CHO and EPOX was continuously followed by FT-RTIR at approximately 780 and 790 cm−1, respectively[15]. The kinetic parameter, Rp/[M]0, was determined from the initial slopes of the irradiation time-conversion curves according to the Eq. (1):
where Rp and [M]0 are the rates of polymerization and the initial monomer concentration, respectively. The conversions were determined from the curves at irradiation time t1 and t2.
Materials
All the chemicals used for synthesis were purchased from Sinopharm Chemical Reagent Co., Ltd., TCI, Alfa Aesar, or J & K Chemical. The reagents were utilized without further purification unless otherwise specified. All the solvents employed for photophysical measurements were Fluka spectroscopic grade. The monomers of cyclohexene oxide (CHO, 98%) and (3, 4-epoxycyclohexane) methyl-3, 4-epoxycyclohexylcarboxylate (EPOX, 98%) were purchased from Sigma-Aldrich with the highest purity and used as received without further purification.
(4-Cyanobenzyl)(4″-(N, N′-diphenylamino)-[1, 1′:4′, 1″-terphenyl]-4-yl)(methyl) sulfoniumhexafluoro-phosphate (PAG-P)
Yield: 50.3%. 1H-NMR (400 MHz, CDCl3, δ): 7.81 (d, J=8.1 Hz, 2H, PhH), 7.74 (d, J=8.1 Hz, 2H, PhH), 7.65 (d, J=8.2 Hz, 2H, PhH), 7.60 (d, J=8.0 Hz, 2H, PhH), 7.52 (d, J=7.6 Hz, 2H, PhH), 7.48 (d, J=8.3 Hz, 2H, PhH), 7.33 (d, J=7.5 Hz, 2H, PhH), 7.26 (t, J=7.3 Hz, 4H, PhH), 7.12 (d, J=7.8 Hz, 6H, PhH), 7.04 (t, J=7.1 Hz, 2H, PhH), 4.95 (m, 2H, CH2), 3.37 (s, 3H, CH3). 13C-NMR (101 MHz, CDCl3, δ): 147.87, 147.82, 147.45, 141.79, 135.73, 133.20, 133.05, 132.24, 131.47, 131.20, 129.38, 129.31, 127.67, 127.33, 124.70, 124.49, 123.47, 123.29, 118.88, 117.74, 114.16, 50.74, 25.92. EI-MS (m/z): calcd. for C39H31F6N2PS, 704.7066, found: 559.2211, [M-PF6-]+.
4-(N, N′-diphenylamine) phenyl-styrene (5)
Potassium tert-butoxide (1.30 g, 11.6 mmol) was added to a solution of methyltriphenylphosphonium bromide (5.80 g, 14.3 mmol) in 50 mL of dry THF at 0 ℃. After the mixture was stirred for 15 min at room temperature, compound 4 (4.15 g, 12.0 mmol) was added at 0 ℃. The mixture was stirred at room temperature for another 3 h and poured into 400 mL of water. The solid product was collected by filtration. The crude product was recrystallized in ethyl alcohol to yield a yellow solid at 89.0%. 1H-NMR (400 MHz, CDCl3, δ): 7.54 (d, J=8.1 Hz, 2H, PhH), 7.47 (t, J=7.4 Hz, 4H, PhH), 7.26 (d, J=4.7 Hz, 4H, PhH), 7.13 (d, J=7.8 Hz, 6H, PhH), 7.03 (t, J=7.3 Hz, 2H, PhH), 6.75 (m, 1H, CH=CH), 5.78 (d, J=17.6 Hz, 1H, CH=CH), 5.26 (d, J=10.9 Hz, 1H, CH=CH).
(E)-(4-(4-(diphenylamino) styryl) phenyl) boronic acid (8)
A solution of compound 7 (4.26 g, 10 mmol) in anhydrous THF (50 mL) was cooled to -78 ℃ and added dropwise with n-BuLi (2.5 mol×L-1 in n-hexane, 8 mL, 20 mmol) at -78 ℃ for 2 h. The reaction mixture was stirred at -78 ℃ for another 1 h. The mixture was immediately added with triisopropyl borate (7 mL, 30 mmol) and allowed to warm at room temperature for 15 h. The reaction was finally quenched with HCl (2.0 mol×L-1, 40 mL), and the mixture was poured into water (100 mL). After extraction with CH2Cl2 (100 mL × 3), the organic layer was washed with brine, dried over MgSO4, and concentrated. Further purification by the silica gel column chromatography (petroleum ether/dichloromethane, 2/1, V/V) produced compound 8 as a yellow powder (2.50 g, 64%). 1H-NMR (400 MHz, DMSO-d6, δ): 8.04 (s, J=7.9 Hz, 2H, OH), 7.78 (d, J=7.9 Hz, 2H, PhH), 7.53 (d, J=2.9 Hz, 2H, PhH), 7.51 (d, J=3.6 Hz, 2H, PhH), 7.32 (t, J=7.8 Hz, 2H, PhH), 7.25 (s, J=16.7 Hz, 2H, PhH), 7.11 (d, J=16.7 Hz, 2H, CH=CH), 7.05 (t, J=7.8 Hz, 6H, PhH), 6.96 (d, J=8.5 Hz, 2H, PhH).
General synthetic procedure for sulfonium salts (PAG)
PAG-P was selected as a sample and prepared in dark because of its high sensitivity to atmospheric light. Pre-P (500 mg, 0.92 mmol) was added to a 100 mL flask with 40 mL of dry dichloromethane. The mixture was stirred at -78 ℃ for 30 min. The solution was added with methyl trifluoromethanesulfonate (124 µL, 1.12 mmol) by using a syringe. After stirring at this temperature for 2 h, the mixture was warmed to room temperature and stirred for another 48 h. Subsequently, dichloromethane was removed, and the residue was dissolved in acetonitrile. This solution was added dropwise with ethyl ether at 10 times the volume to facilitate crystal formation. The solid was collected by filtration, washed with ethyl ether, and dissolved in acetonitrile for another round of crystal formation by dropping ethyl ether at 10 times the volume. Anion exchange of CF3SO3- by PF6- was performed with the method described by Zhou et al[25]. Briefly, 50 mL of fresh aqueous KPF6 (10 eq.) solution was added to the prepared PAG solution in 20 mL of acetone. The mixture was stirred for 2 h at room temperature in dark. The solid was collected by filtration and redissolved in 10 mL of acetone. The anion-exchange process was performed three times to obtain the final product.
4-(4-(2-(4′-(Diphenylamino)-[1, 1′-biphenyl]-4-yl) vinyl) phenyl)-4-cyanobenzyl sulfane (Pre-PS)
4-(N, N-diphenylamine) phenyl-styrene (5) (3.47 g, 10 mmol), compound 1 (3.04 g, 10 mmol), and Pd (OAc)2(22.4 mg, 0.1 mmol) were mixed with 30 mL of triethanolamine in a flask. The mixture was stirred for 12 h in an oil bath at 120 ℃ under nitrogen atmosphere, cooled to room temperature, and poured into 200 mL of water. The mixture was extracted with dichloromethane (100 mL x 3), and the organic layer was washed three times with brine, dried with anhydrous MgSO4, and filtered over a short pad of silica gel to remove the Pd catalyst. The solvent was removed to obtain a green-yellow powder as the crude product. This product was purified by the silica gel column chromatography (petroleum ether/dichloromethane, 10/1, V/V), and 3.46 g (70%) of Pre-PS was obtained. 1H-NMR (400 MHz, CDCl3, δ): 7.54 (d, 6H, PhH), 7.48 (d, 2H, PhH), 7.39 (d, 2H, PhH), 7.33 (d, 2H, PhH), 7.25 (m, 6H, PhH), 7.13 (d, 6H, PhH), 7.00 (m, 4H, PhH, CH=CH), 4.09 (s, 2H, CH2). 13C-NMR (101 MHz, CDCl3, δ): 147.65, 147.36, 143.38, 140.00, 136.46, 135.58, 134.31, 133.81, 132.31, 131.07, 129.54, 129.34, 128.83, 127.54, 127.39, 127.04, 126.83, 124.53, 123.82, 123.07, 118.78, 111.04, 39.23.
(E)-(4-cyanobenzyl)(4′-(4-(diphenylamino) styryl)-[1, 1′-biphenyl]-4-yl)(methyl) sulfonium hexafluorophosphate (PAG-SP)
Yield: 68.0%. 1H-NMR (400 MHz, CD3CN, δ): 7.92 (d, J=8.6 Hz, 2H, PhH), 7.75 (d, J=8.7 Hz, 2H, PhH), 7.73-7.64 (m, 6H, PhH), 7.47 (d, J=8.6 Hz, 2H, PhH), 7.36 (d, J=8.3 Hz, 2H, PhH), 7.33-7.23 (m, 5H, PhH, CH=CH), 7.16-7.03 (m, 7H, PhH, CH=CH), 6.98 (d, J=8.5 Hz, 2H, PhH), 4.94-4.70 (m, 2H, CH2), 3.21 (s, 3H, CH3). 13C-NMR (101 MHz, CD3CN, δ): 147.13, 146.47, 138.48, 136.16, 132.78, 132.20, 131.21, 131.05, 129.17, 129.02, 128.44, 127.37, 127.32, 126.69, 124.25, 123.12, 122.67, 119.42, 117.59, 113.30, 49.85, 24.39. EI-MS (m/z): calcd. for C41H33F6N2PS, 730.7433, found: 585.2363, [M-PF6-]+.
4-(4-(N, N′-diphenylamino) styryl) phenyl 4-cyanobenzyl sulfane (Pre-S)
Pre-S was prepared with the same method of Pre-PS by the Heck reaction from compounds 9 and 1. The product can be purified by the silica gel column chromatography (petroleum ether-dichloromethane, 10/1, V/V) to obtain a yield of 78.8%. 1H-NMR (400 MHz, CDCl3, δ): 7.55 (d, 2H, PhH), 7.38 (m, 6H, PhH), 7.24 (m, 5H, PhH), 7.05 (m, 10H, PhH, CH=CH), 6.91 (d, 1H, CH=CH), 4.09 (s, 2H, CH2). 13C-NMR (101 MHz, CDCl3, δ): 147.65, 147.54, 143.52, 136.84, 133.33, 132.37, 131.28, 131.12, 129.61, 129.42, 128.82, 127.50, 126.90, 125.94, 124.68, 123.47, 123.25, 118.88, 111.05, 39.40.
4′-(N, N′-diphenylamino) phenyl-benzaldehyde (4)
A solution of compound 2 (4.00 g, 10 mmol) in anhydrous THF (50 mL) was cooled to −78 ℃ and added dropwise with n-BuLi (6 mL, 2.5 mol×L-1 in hexane, 15 mmol). The mixture was stirred for another 15 min before the dropwise addition of dry DMF (3 mL, 17.5 mmol). The reaction mixture was stirred for 18 h before warming to room temperature. The reaction was quenched by addition of saturated NH4Cl (aq.), followed by water. The product was extracted into diethyl ether (50 mL x 2), and the combined organic layers were dried (MgSO4) and concentrated. Purification by the chromatography on silica with 5% to 10% ethyl acetate in hexane yielded compound 4 (2.80 g, 80% yield). 1H-NMR (400 MHz, CDCl3, δ): 10.01 (s, 1H, CHO), 7.91 (d, J=8.1 Hz, 2H, PhH), 7.71 (d, J=8.1 Hz, 2H, PhH), 7.51 (d, J=8.6 Hz, 2H, PhH), 7.27 (d, J=7.9 Hz, 4H, PhH), 7.14 (d, J=7.6 Hz, 6H, PhH), 7.06 (t, J=7.3 Hz, 2H, PhH).
(E)-(4-cyanobenzyl)(4-(4-(N, N′-diphenylamino) styryl) phenyl)(methyl) sulfonium hexafluorophosphate (PAG-S)
Yield: 52.2%. 1H-NMR (CDCl3, δ): 7.72 (d, 2H, PhH), 7.66 (d, 2H, PhH), 7.55 (d, 2H, PhH), 7.39 (d, 4H, PhH), 7.30 (t, 4H, PhH), 7.14 (d, 4H, PhH), 7.10-7.03 (m, 5H, PhH, CH=CH), 6.92 (d, 1H, CH=CH), 5.20 (m, 2H, CH2), 3.47 (s, 3H, CH3). 13C-NMR (CDCl3, δ): 148.49, 147.19, 144.55, 133.05, 132.91, 131.52, 131.37, 129.92, 129.58, 128.21, 128.02, 124.90, 123.88, 123.78, 122.30, 119.49, 118.57, 117.98, 113.45, 50.05, 25.04. EI-MS (m/z): calcd. for C35H29F6N2PS, 654.1733, found: 509.2363, [M-PF6-]+.
(E)-4-((4′-(N, N′-4-(diphenylamino) styryl)-[1, 1′-biphenyl]-4-yl 4-cyanobenzyl sulfane (Pre-SP)
Pre-SP was prepared by the same method as Pre-P from 8 and 1. The crude product was purified by the silica gel (200-300 mesh) column chromatography with petroleum ether/dichloromethane (5/1, V/V) as an eluent to obtain Pre-SP as a pale yellow powder with an isolation yield of 65%. 1H-NMR (400 MHz, CDCl3, δ): 7.56 (d, J=8.6 Hz, 2H, PhH), 7.55 (s, 4H, PhH), 7.50 (d, J=8.2 Hz, 2H, PhH), 7.38 (t, J=9.4 Hz, 4H, PhH), 7.32 (d, J=8.2 Hz, 2H, PhH), 7.25 (d, J=7.4 Hz, 4H, PhH), 7.11 (d, J=7.9 Hz, 4H, PhH), 7.05 (dd, J1=13.4 Hz, J2=5.0 Hz, 5H, CH=CH, PhH), 7.00 (d, J=10.8 Hz, 1H, CH=CH), 4.12 (s, 2H, CH2). 13C-NMR (101 MHz, CDCl3, δ): 147.53, 143.37, 139.51, 138.75, 137.06, 133.83, 132.34, 131.34, 131.06, 129.57, 129.37, 128.51, 127.47, 127.38, 127.10, 126.86, 126.32, 124.59, 123.52, 123.16, 118.80, 111.06, 39.15.
4-(4″-(N, N′-diphenylamino)-[1, 1′:4′, 1″-terphenyl]-4-yl)-4-cyanobenzyl sulfane (Pre-P)
A mixture of compound 1 (3.04 g, 10 mmol), tetrakis (triphenylphosphine) palladium (0) (585 mg, 0.5 mmol), compound 3 (3.65 g, 10 mmol), 2 mol×L-1 K2CO3 (20 mL), and THF (80 mL) was heated at 80 ℃ overnight with magnetic stirring under a nitrogen atmosphere. After cooling to room temperature, the reaction mixture was poured into water and extracted with dichloromethane (50 mL × 3). The combined organic layer was dried with anhydrous Na2SO4, and evaporated to dryness. The crude product was purified by the silica gel (200-300 mesh) column chromatography with 5:1 petroleum ether-dichloromethane as an eluent to obtain Pre-P as a pale yellow powder with an isolation yield of 68%.
(E)-(4-cyanobenzyl)(4-(2-(4′-(diphenylamino)-[1, 1′-biphenyl]-4-yl) vinyl) phenyl)(methyl) sulfonium hexafluorophosphate (PAG-PS)
Yield: 45.7%. 1H-NMR (400 MHz, CD3CN, δ): 7.80 (d, J=8.6 Hz, 2H, PhH), 7.71 (d, J=8.3 Hz, 2H, PhH), 7.69-7.64 (m, 6H, PhH), 7.58 (d, 2H, PhH), 7.45 (d, 1H, CH=CH), 7.38-7.23 (m, 7H, PhH, CH=CH), 7.08 (m, 8H, PhH), 4.93-4.67 (m, 2H, CH2), 3.19 (s, 3H, CH3). 13C-NMR (101 MHz, CD3CN, δ): 147.34, 147.20, 144.03, 140.21, 134.67, 132.81, 132.77, 132.28, 131.17, 130.97, 129.18, 128.01, 127.39, 127.21, 126.38, 125.39, 124.22, 123.10, 122.98, 118.64, 117.59, 113.28, 49.87, 24.52. EI-MS (m/z): calcd. for C41H33F6N2PS, 730.7433, found: 585.2363, [M-PF6-]+.
RESULTS AND DISCUSSION
Photopolymerization
A relevant criterion for evaluating the relative performances of PAGs is to determine the effective efficiency for H+ photogeneration. This fundamental criterion can be obtained by multiplying ΦH+ with ελ. The latter parameter represents the photon-induced ability to generate excited species upon excitation at a special wavelength, whereas the first factor indicates the efficiency to produce H+ from the excited species. The corresponding values are reported for ΦH+·ε365 nm in Table 1. The efficiency of PAG-S was lowest because of its small ΦH+ at 365 nm, whereas PAG-P had the best efficiency. To corroborate these preliminary trends with the photoinitiating efficiencies of PAG, the cationic photopolymerizations were performed on CHO via the excitation at 365 nm[25, 35, 36]. Figure 5 and Table 1 show the FT-RTIR kinetic curves and the rate of polymerization Rp/[M]0during the irradiation at 365 nm for the formulations containing PAGs (1 wt%), respectively. The photoinitiation efficiencies were clearly correlated with ΦH+·ε365nm. PAG-P underwent rapid photopolymerization with an epoxide conversion rate of 70% after 50 s of irradiation. PAG-SP exhibited a comparable kinetic curve, but with a higher conversion at 200 s than PAG-P. PAG-P had a final conversion of over 80% after 300 s of irradiation.
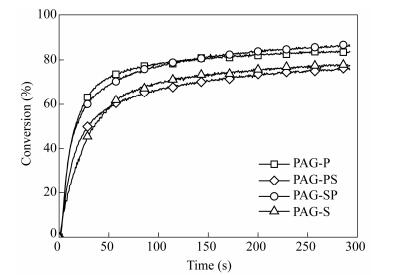 Figure 5.
Conversion versus time curves for the cationic photopolymerization of CHO containing PAGs (1 wt%) (λexc: 365 nm, irradiance: 5 mW×cm-2)
Figure 5.
Conversion versus time curves for the cationic photopolymerization of CHO containing PAGs (1 wt%) (λexc: 365 nm, irradiance: 5 mW×cm-2)
The photopolymerization of CHO in the presence of PAG-S (1 wt%) in air was also conducted by exposure to 385-425 nm LEDs because the reaction showed considerable extinction coefficients in this range (Fig. 1 and Table 1). PAG-S can initiate the polymerization of CHO at all wavelengths and the conversion profiles for the reaction are similar with that at 365 nm (data were not shown). The conversion at t=5 min is depicted in Fig. 6(a). Interestingly, the polymerization conversion was improved upon addition of commercial diphenyl-iodoniumhexafluorophosphate (Iod) which was always used in the photosensitizing systems for the visible light photopolymerization[37] (Fig. 6a). The conversion of the two-component PAG-S/Iod systems exceeded 85%-90%. The satisfactory performance of the two-component system at 365-425 nm could be attributed to the contribution of photoinduced electron transfer from the excited precursor analogue (byproducts shown in Scheme 3) as a sensitizer to the Iod, which was discussed in our previous report[29]. According to the photolysis mechanism shown in Scheme 3, byproducts are present in the photoinitiating systems; thus, the photo-sensitization process occurs in the present work.
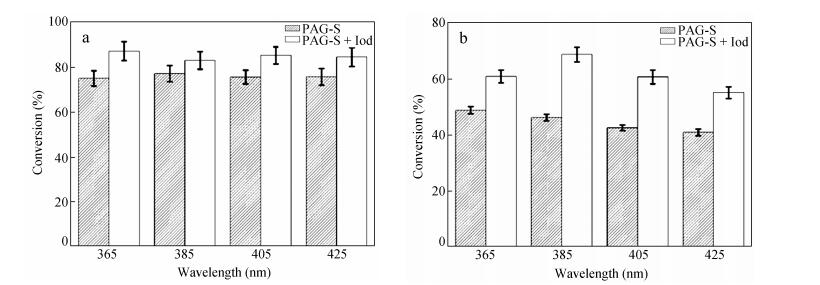 Figure 6.
(a) Photopolymerization conversions of CHO in air and in the presence of different initiating systems (1 wt% PAG-S, 3 wt% Iod) at different LED irradiation (5 mW×cm-2); (b) photopolymerization conversions of EPOX in air and in the presence of different initiating systems (1 wt% PAG-S, 3 wt% Iod) at different LED irradiation (10 mW×cm-2)
Figure 6.
(a) Photopolymerization conversions of CHO in air and in the presence of different initiating systems (1 wt% PAG-S, 3 wt% Iod) at different LED irradiation (5 mW×cm-2); (b) photopolymerization conversions of EPOX in air and in the presence of different initiating systems (1 wt% PAG-S, 3 wt% Iod) at different LED irradiation (10 mW×cm-2)
PAG-S and PAG-S/Iod can efficiently initiate the polymerization of EPOX with a relative conversion of ca. 45-70%. They both can be considered as the excellent photoinitiating systems for this viscous monomer[38]. The related polymerization conversions are provided in Fig. 6(b). The exposure wavelengths red-shifted to the visible range (425 nm), and the two-component systems still exhibited good conversions compared with PAG-S alone (Fig. 6b). This result is highly dependent on the photosensitizing process. The final tack-free coatings are obtained within 5 min of LED irradiation at different wavelengths.
Photochemical and Photophysical Properties of the PAGs
The absorption spectra of all PAGs in acetonitrile are shown in Fig. 1. The maxima for PAGs are located in the UV-Vis range (Table 1) at λmax(PAG-P)=360 nm (ε360 nm, ~23500 L×mol-1×cm-1), λmax(PAG-PS)=380 nm (ε380 nm, ~24500 L×mol-1×cm-1), λmax(PAG-SP)=390 nm (ε390 nm, ~26800 L×mol-1×cm-1), and λmax(PAG-S)=400 nm (ε400 nm, ~31000 L×mol-1×cm-1). Compared with the spectrum of PAG-P, other PAGs display the gradually increased red-shift absorbance and higher ε values because of their stronger conjugated effect[20]. The excellent absorption properties of PAGs are associated with the large delocalization of the highest occupied molecular orbit (HOMO) and the lowest unoccupied molecular orbit (LUMO), which are involved in the lowest energy transitions[20, 29]. This result is further confirmed by DFT calculations, as shown in Fig. 2. The HOMOs of sulfonium salts are mainly localized on the donor of triphenylamine and conjugated system moieties. However, the distribution of electron clouds is obviously different in HOMOs. The delocalization of electron clouds in the conjugated systems are consistent with the absorption wavelength of PAGs, e.g., the HOMO orbit of PAG-S was extended to the whole conjugated system, but the corresponding orbit in PAG-P was just extended to the middle benzene ring, thereby indicating that the distribution of the frontier orbits has a decisive influence on the absorption properties of the molecules. The theoretical calculations provide an easy method to predict the photochemical properties in these kinds of sulfonium PAGs. The theoretical calculation results can also be proved by 1H-NMR data, as shown in Fig. 3. The chemical shifts in 1H-NMR provide the useful information on the electromagnetic shielding in a molecule. In the present study, the signals from the protons around the sulfonium salt in PAG-P shifted to a low field as compared with those for other PAGs. The excited states, e.g., LUMOs, are mainly distributed throughout the sulfonium moieties. Saeva proved that the photoreaction for arylsulfonium salts occurred by direct excitation of a π-σ transition (S-C bond) or by excitation of a π-π transition, which was followed by the intramolecular electron transfer to the σ on the S-C bond[17, 32, 33]. In both cases, the subsequent homolytic cleavage of the S-C bond led to the formation of a radical cation on the sulfur-containing fragment and a neutral radical on the carbon fragment. The results obtained from the theoretical calculations confirm that the S-C σ bond (face-to-face) exists in the LUMO orbits of PAGs, as shown in Fig. 2.
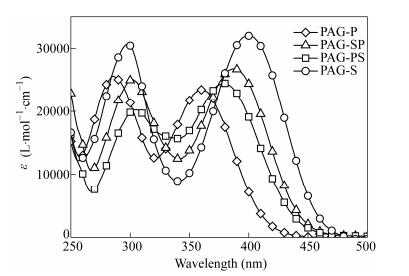 Figure 1.
UV-Vis spectra of PAG-P, PAG-SP, PAG-PS, and PAG-S in acetonitrile
Figure 1.
UV-Vis spectra of PAG-P, PAG-SP, PAG-PS, and PAG-S in acetonitrile
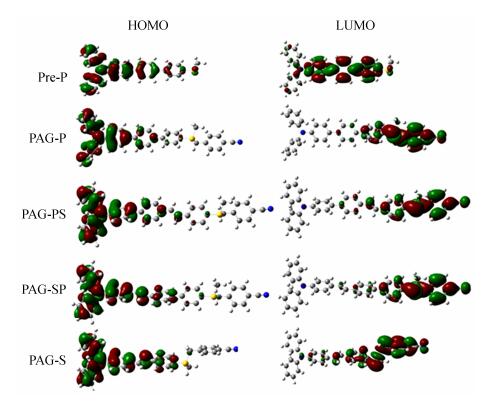 Figure 2.
HOMO and LUMO of Pre-P and four PAGs at the DFT B3LYP/6-31 G (d) level
Figure 2.
HOMO and LUMO of Pre-P and four PAGs at the DFT B3LYP/6-31 G (d) level
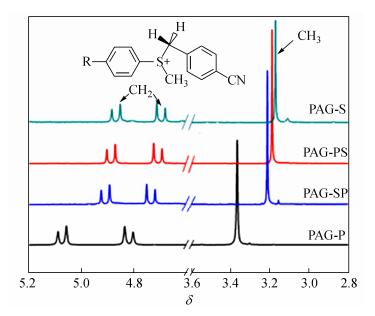 Figure 3.
1H-NMR spectra of four PAGs in CD3CN
Figure 3.
1H-NMR spectra of four PAGs in CD3CN
 Table 1.
Optical data and parameters for the precursors and PAGs in acetonitrile
Table 1.
Optical data and parameters for the precursors and PAGs in acetonitrile
Compound λabs (nm) εmax (L×mol-1×cm-1) λem (nm) Фfluo ФH+ a ФH+·ε365 nm (L×mol-1×cm-1) Rp/[M]0(s-1) PAG-P 360 23500 - - 0.58 13400 4.23 PAG-PS 380 24500 - - 0.50 10100 2.82 PAG-SP 390 26800 - - 0.48 10500 4.12 PAG-S 400 31000 - - 0.32 5800 2.10 Pre-P 344 29800 449 0.89 - Pre-PS 367 39800 503 0.41 - Pre -SP 378 45300 489 0.57 - Pre -S 375 32300 480 0.31 - a Irradiated at 365 nm; uncertainty of 10% Table 1. Optical data and parameters for the precursors and PAGs in acetonitrileThe molar extinction coefficients of all PAGs were smaller than those of their corresponding precursors, as shown in Table 1. The frontier orbits of the precursor (Pre-P as sample) and all PAGs were calculated at the DFT level and are presented in Fig. 2. In the precursor, a very slight charge transfer from the amino group to the conjugated moieties (HOMO-LUMO) was observed in the entire compound structure (π-π type). However, the HOMO-LUMO orbits for the PAGs showed a charge transfer from the amino groups to the sulfonium, and the π-σ type transition overlapped with the π-π transition[3]. The former could not be clearly observed because the chemical reactions involved a change in the precursors to the sulfonium salts, which altered the nature of electron transitions. Table 2 shows the calculated energies of the electronic transitions of PAGs with their corresponding oscillator strengths, where the longest wavelength electronic transition was strongly allowed (f > 1). The π-σ type transition had smaller molar extinction coefficients (ε) than the π-π type transition. Thus, the ε of PAGs was lower than that of their precursors, and the π-σ type transition implied that the PAGs had good ΦH+ performance.
Table 2.
TD-DFT and experimental electronic transitions of PAGs
Transitions PAG-P PAG-PS PAG-SP PAG-S Eth (eV) f Eth (eV) f Eth (eV) f Eth (eV) f S0→S1 3.88 1.56 3.20 2.36 3.17 2.30 3.08 1.93 S0→S2 4.45 0.03 4.06 0.04 4.14 0.02 4.38 0.03 S0→S3 4.58 0.31 4.39 0.04 4.37 0.03 4.43 0.01 S0→S4 4.61 0.04 4.46 0.28 4.56 0.25 4.63 0.23 S0→S5 4.92 0.51 4.58 0.26 4.69 0.03 4.74 0.02 Table 2. TD-DFT and experimental electronic transitions of PAGsThe photoexcitation process of PAGs was examined. The dark stability was checked, and the results showed that the stability of all the remaining ratios were higher than 99%. These compounds are stable under cold and dark conditions. Detailed photolysis at an excitation wavelength of 365 nm was performed and analyzed by UV-Vis spectroscopy, as shown in Fig. 4. Two kinds of photolysis curves were observed. For PAG-P and PAG-PS, the absorption peaks blue-shifted, and the increased intensity suggested that the byproducts were the precursors. For PAG-SP and PAG-S, the decreased absorption of the strongest transition suggested that the conjugated systems were destroyed. Based on the report of Saeva and our previous results[3, 21], a possible photolysis mechanism is proposed and shown in Scheme 3. The 4-cyanobenzyl radical (Bz·) attacks the conjugated systems to form the precursor and H+. If Bz· reacts with H2O and the solvent, or initiates the polymerization of monomers[34], more precursor can be reproduced and it can also be used as a sensitizer during the photopolymerization[29], which will be discussed thereafter.
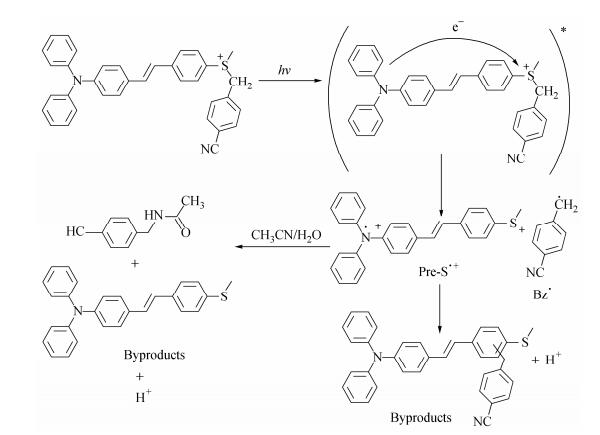 Figure Scheme 3.
Proposed mechanism of sulfonium photolysis
Figure Scheme 3.
Proposed mechanism of sulfonium photolysis
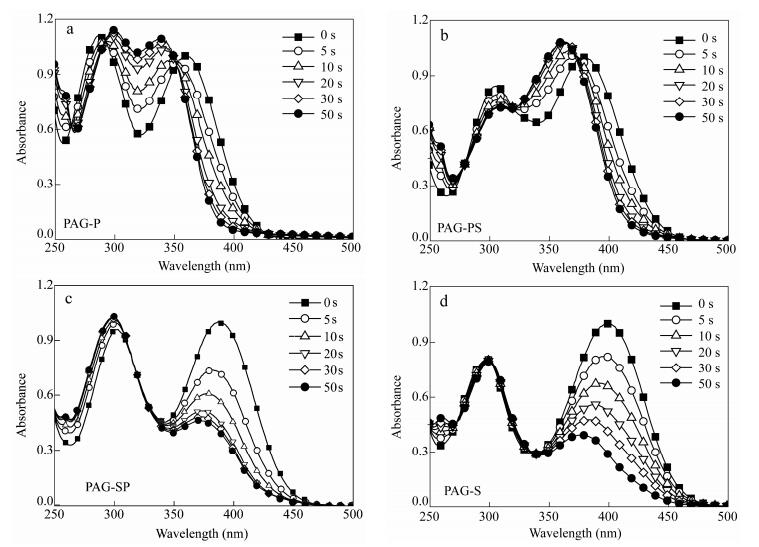 Figure 4.
UV-Vis spectra of four PAGs in acetonitrile under LED irradiation at 365 nm (0.4 mW×cm-2): (a) PAG-P, (b) PAG-PS, (c) PAG-SP and (d) PAG-S
Figure 4.
UV-Vis spectra of four PAGs in acetonitrile under LED irradiation at 365 nm (0.4 mW×cm-2): (a) PAG-P, (b) PAG-PS, (c) PAG-SP and (d) PAG-S
The quantum yields for photoacid generation (ΦH+) in Table 1 were measured in acetonitrile, with RhB as an acid indicator via the method reported by Scaiano et al[26]. The efficiency of acid generation was strongly dependent on the conjugated systems[18, 19]. As proved by Fig. 4, when Bz· attacks phenyl rings, e.g., in PAG-P and PAG-PS, the radical has better performance than when it reacts with styryl groups, e.g., in PAG-SP and PAG-S. The smallest ΦH+ was 0.32 for PAG-S, but this value was still higher when compared with those reported for PAGs having higher absorption wavelength[18, 19]. The use of styryl and phenyl as conjugated systems was beneficial to improve the performance of PAGs. The appropriate selection and position of styryl and phenyl groups are crucial in obtaining good ΦH+ values.
Synthesis
Four sulfonium salts were prepared from 4-substituted triphenylamine with 4-bromophenyl 4-cyanobenzyl sulfane via the Suzuki and Heck reactions (Scheme 2)[21, 23, 28]. Pd-catalyzed couplings of the triphenylamine-containing conjugated system were used because the triphenylamine group has been proven to be a good substituent in sulfonium salts[31]. The 4-cyanobenzyl moiety was chosen because of its better leaving propensity[20]. Finally, the alkylation of precursors with CF3SO3CH3 gave different conjugated systems, in which the sulfoniumtriflate salts; non-nucleophilic anions, such as PF6-, were exchanged to benefit the polymerization reactions[31].
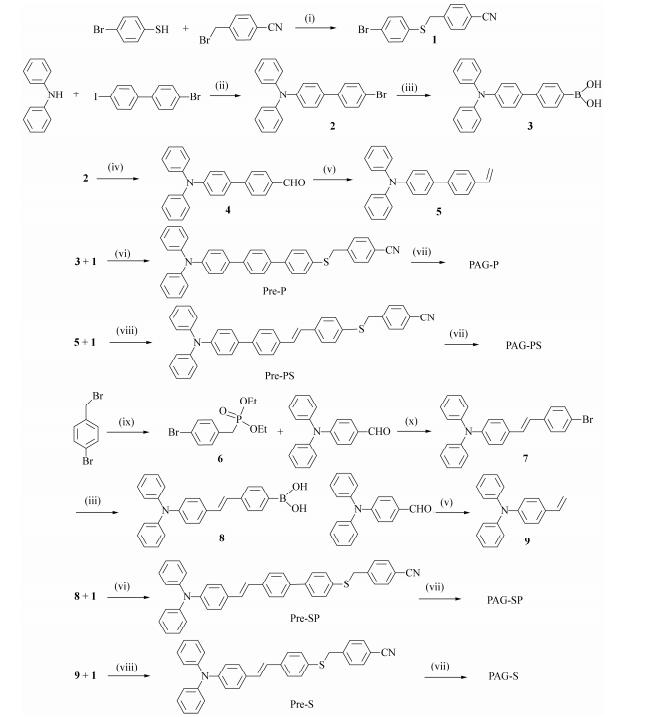 Figure Scheme 2.
The synthetic routes to PAGs: (i) K2CO3, acetone, reflux; (ii) Cu, K2CO3, 200 ℃, 1, 2-dichlorobenzene; (iii) n-BuLi, triisopropyl borate, THF, -78 ℃; (iv) n-BuLi, THF, -78 ℃, N2, DMF; (v) Ph3PCH3Br, t-BuOK, THF, 0 ℃, r.t.; (vi) Pd (PPh3)4, K2CO3, THF; (vii) CF3SO3CH3, CH2Cl2, KPF6 (water/acetone); (viii) Pd (OAc)2, N (CH2CH2OH)3, N2, 120 ℃; (ix) (EtO)3P, reflux; and (x) t-BuOK, THF, r.t.
Figure Scheme 2.
The synthetic routes to PAGs: (i) K2CO3, acetone, reflux; (ii) Cu, K2CO3, 200 ℃, 1, 2-dichlorobenzene; (iii) n-BuLi, triisopropyl borate, THF, -78 ℃; (iv) n-BuLi, THF, -78 ℃, N2, DMF; (v) Ph3PCH3Br, t-BuOK, THF, 0 ℃, r.t.; (vi) Pd (PPh3)4, K2CO3, THF; (vii) CF3SO3CH3, CH2Cl2, KPF6 (water/acetone); (viii) Pd (OAc)2, N (CH2CH2OH)3, N2, 120 ℃; (ix) (EtO)3P, reflux; and (x) t-BuOK, THF, r.t.
CONCLUSIONS
The photoreactivity of a new series of sulfonium-based PAGs was evaluated, with different styryl and phenyl groups as the π-conjugated systems. The length and configuration of the conjugated system had a significant effect on the linear absorption and caused a significant increase in the quantum yield for acid generation (ΦH+=0.58 for a triphenyl-containing PAG, PAG-P). Moreover, the extended and twisted conjugated systems shifted the absorption peaks to the near-UV range and increased the molar extinction coefficients. From a practical perspective, we may conclude that the photoinitiating performances of the PAGs and the properties of the final photopolymers are very well correlated with the relative reactivities of these photoinitiators. Finally, we show that this new series of PAGs is of potential interest for the microfabrication technologies and can be employed in i-line chemically-amplified photoresists.
-
-
[1]
Crivello, J.V. and Lam, J.H.W., Macromolecules, 1977, 10(6):1307 doi: 10.1021/ma60060a028
-
[2]
Beak, P. and Sullivan, T.A., J. Am. Chem. Soc., 1982, 104(16):4450 doi: 10.1021/ja00380a020
-
[3]
Saeva, F.D. and Morgan, B.P., J. Am. Chem. Soc., 1984, 106(15):4121 doi: 10.1021/ja00327a010
-
[4]
Fouassier, J.P. and Lalevée, J., "Photoinitiator for polymer synthesis-scope, reactivity, and efficiency", Wiley-VCH Berlag GmbH & Co. KGaA, Weinheim, 2012
-
[5]
Davidson, S., "Exploring the science, technology and application of UV and EB curing", Sita Technolgy Ltd, London, 1999
-
[6]
Fouassier, J.P., "Photoinitiator, photopolymerization and photocuring:fundamentals and applications", Hanser Publishers, New York, 1995
-
[7]
Cordon, C. and Miller, C., "UV-LED:presented by RadTech-the association for UV & EB technology", RadTech International, Bethesda, 2013
-
[8]
Liu, G., Zhu, X., Xu, B., Qian, X., Song, G. and Nie, J., J. Appl. Polym. Sci., 2013, 130(5):3698 doi: 10.1002/app.39612
-
[9]
Sui, X., Feng, X., di Luca, A., van Blitterswijk, C.A., Moroni, L., Hempenius, M.A. and Vancso, G.J., Polym. Chem., 2013, 4(2):337 doi: 10.1039/C2PY20431B
-
[10]
Tehfe, M.A., Dumur, F., Graff, B., Morlet-Savary, F., Fouassier, J.P., Gigmes, D. and Lalevée, J., Macromolecules, 2012, 45(21):8639 doi: 10.1021/ma301931p
-
[11]
Walsh, Z., Levkin, P.A., Jain, V., Paull, B., Svec, F. and Macka, M., J. Sep. Sci., 2010, 33(1):61 doi: 10.1002/jssc.v33:1
-
[12]
Xiao, P., Frigoli, M., Dumur, F., Graff, B., Gigmes, D., Fouassier, J.P. and Lalevée, J., Macromolecules, 2014, 47(1):106 doi: 10.1021/ma402196p
-
[13]
Dietlin, C., Schweizer, S., Xiao, P., Zhang, J., Morlet-Savary, F., Graff, B., Fouassier, J.P. and Lalevée, J., Polym. Chem., 2015, 6(21):3895 doi: 10.1039/C5PY00258C
-
[14]
Tehfe, M.A., Dumur, F., Xiao, P., Delgove, M., Graff, B., Fouassier, J.P., Gigmes, D. and Lalevée, J., Polym. Chem., 2014, 5(2):382 doi: 10.1039/C3PY00922J
-
[15]
Xiao, P., Hong, W., Li, Y., Dumur, F., Graff, B., Fouassier, J.P., Gigmes, D. and Lalevée, J., Polym. Chem., 2014, 5(7):2293 doi: 10.1039/c3py01599h
-
[16]
Zhang, J., Zivic, N., Dumur, F., Xiao, P., Graff, B., Gigmes, D., Fouassier, J.P. and Lalevée, J., J. Polym. Sci., Part A:Polym. Chem., 2015, 53(3):445 doi: 10.1002/pola.v53.3
-
[17]
Saeva, F.D., Garcia, E. and Martic, P.A., J. Photochem. Photobiol. A-Chem., 1995, 86(1-3):149 doi: 10.1016/1010-6030(94)03923-I
-
[18]
Zhou, W.H., Kuebler, S.M., Braun, K.L., Yu, T.Y., Cammack, J.K., Ober, C.K., Perry, J.W. and Marder, S.R., Science, 2002, 296(5570):1106 doi: 10.1126/science.296.5570.1106
-
[19]
Yanez, C.O., Andrade, C.D. and Belfield, K.D., Chem. Commun., 2009, 0(7):827
-
[20]
Xia, R., Malval, J.P., Jin, M., Spangenberg, A., Wan, D., Pu, H., Vergote, T., Morlet-Savary, F., Chaumeil, H., Baldeck, P., Poizat, O. and Soppera, O., Chem. Mater., 2012, 24(2):237 doi: 10.1021/cm2030075
-
[21]
Jin, M., Hong, H., Xie, J., Malval, J.P., Spangenberg, A., Soppera, O., Wan, D., Pu, H., Versace, D.L., Leclerc, T., Baldeck, P., Poizat, O. and Knopf, S., Polym. Chem., 2014, 5(16):4747 doi: 10.1039/C4PY00424H
-
[22]
De Waele, V., Hamm, M., Vergote, T., Chaumeil, H., Jin, M., Malval, J.P., Baldeck, P. and Poizat, O., Chem. Mater., 2015, 27(5):1684 doi: 10.1021/cm504474g
-
[23]
Jin, M., Wu, X., Xie, J., Malval, J.P. and Wan, D., RSC Adv., 2015, 5(68):55340 doi: 10.1039/C5RA11350D
-
[24]
Saeva, F.D., Breslin, D.T. and Luss, H.R., J. Am. Chem. Soc., 1991, 113(14):5333 doi: 10.1021/ja00014a028
-
[25]
Zhou, W., Kuebler, S.M., Carrig, D., Perry, J.W. and Marder, S.R., J. Am. Chem. Soc., 2002, 124(9):1897 doi: 10.1021/ja011186k
-
[26]
Pohlers, G., Scaiano, J.C. and Sinta, R., Chem. Mater., 1997, 9(12):3222 doi: 10.1021/cm970587p
-
[27]
Frisch, M.J., "Gaussian 09, Revision B.01.", Gaussian Inc, Wallingford, 2009
-
[28]
Fang, B., Jin, M., Wu, X., Zhang, Y. and Wan, D., Dyes Pigment., 2016, 126:54 doi: 10.1016/j.dyepig.2015.11.012
-
[29]
Jin, M., Xie, J., Malval, J.P., Spangenberg, A., Soppera, O., Versace, D.L., Leclerc, T., Pan, H., Wan, D., Pu, H., Baldeck, P., Poizat, O. and Knopf, S., J. Mater. Chem. C, 2014, 2(35):7201 doi: 10.1039/C4TC00706A
-
[30]
Jin, M., Xu, H., Hong, H., Malval, J.P., Zhang, Y., Ren, A., Wan, D. and Pu, H., Chem. Commun., 2013, 49(76):8480 doi: 10.1039/c3cc43018a
-
[31]
Zhou, W.H., Kuebler, S.M., Carrig, D., Perry, J.W. and Marder, S.R., J. Am. Chem. Soc., 2002, 124(9):1897 doi: 10.1021/ja011186k
-
[32]
Saeva, F.D., Breslin, D.T. and Luss, H.R., J. Am. Chem. Soc., 1991, 113(14):5333 doi: 10.1021/ja00014a028
-
[33]
Wang, X.Z., Saeva, F.D. and Kampmeier, J.A., J. Am. Chem. Soc., 1999, 121(18):4364 doi: 10.1021/ja982932x
-
[34]
Park, J., Kihara, N., Ikeda, T. and Endo, T., Macromolecules, 1997, 30(11):3414 doi: 10.1021/ma9610222
-
[35]
Kuebler, S.M., Braun, K.L., Zhou, W., Cammack, J.K., Yu, T., Ober, C.K., Marder, S.R. and Perry, J.W., J. Photochem. Photobiol., A, 2003, 158(2-3):163 doi: 10.1016/S1010-6030(03)00030-3
-
[36]
Yagci, Y., Jockusch, S. and Turro, N.J., Macromolecules, 2010, 43(15):6245 doi: 10.1021/ma1007545
-
[37]
Tehfe, M.A., Dumur, F., Contal, E., Graff, B., Morlet-Savary, F., Gigmes, D., Fouassier, J.P. and Lalevée, J., Polym. Chem., 2013, 4(5):1625 doi: 10.1039/C2PY20950K
-
[38]
Tehfe, M.A., Lalevée, J., Morlet-Savary, F., Graff, B., Blanchard, N. and Fouassier, J.P., ACS Macro Lett., 2011, 1(1):198
-
[1]
-

 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 1001
- HTML全文浏览量: 25
 下载:
下载:
