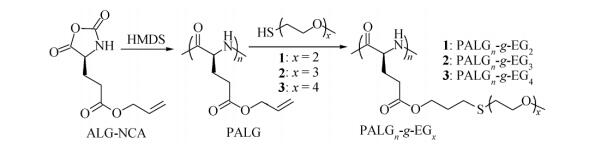
 Figure Scheme 1.
The synthetic route to PALGn-g-EGx by a combination of ROP of ALG-NCA with thiol-ene photo addition
Figure Scheme 1.
The synthetic route to PALGn-g-EGx by a combination of ROP of ALG-NCA with thiol-ene photo addition
Citation:
Xiao-hui Fu, Yi-nan Ma, Jing Sun, Zhi-bo Li. Thermal and Redox Dual Responsive Poly (L-glutamate) with Oligo (ethylene glycol) Side-chains[J]. Chinese Journal of Polymer Science,
2016, 34(12): 1436-1447.
doi:
10.1007/s10118-016-1861-x

Thermal and Redox Dual Responsive Poly (L-glutamate) with Oligo (ethylene glycol) Side-chains
English
Thermal and Redox Dual Responsive Poly (L-glutamate) with Oligo (ethylene glycol) Side-chains
Abstract:
A series of poly (L-glutamate) s grafted with oligo (ethylene glycol) (OEG) side-chains through the thioether linkages (PALGn-g-EGx, x=2, 3 and 4) were prepared by ring-opening polymerization (ROP) of γ-allyl-L-glutamate N-carboxyanhydride (ALG-NCA) and thiol-ene photoaddition. The chemical structures and physical properties were characterized by 1H-NMR, Fourier transform infrared (FTIR), circular dichroism (CD), etc. The PALGn-g-EGx samples with x=3 and 4 displayed lower critical solution temperature (LCST) in water due to the presence of OEG units. The clouding point (CP) of polypeptides can be finely tuned by changing the side chain structures, molecular weights and sample concentrations. In addition, the thioether linkages in the side chains offer additional redox-responsive properties. The influence of both OEG units and thioether linkages on the LCST behavior was systematically investigated. This work provides an efficient way to prepare multi-stimuli responsive materials with highly tunable properties.
-
INTRODUCTION
Stimuli-responsive polymers have attracted tremendous attention for their promising applications in nanotechnology and biotechnology[1-3]. Particularly, polypeptide-based stimuli-responsive materials possess many intriguing features such as excellent biocompatibility, biodegradability and unique assembling property[4-6]. Polypeptides can form highly ordered secondary structures (e.g., α-helices and β-sheets) that can respond to a variety of external stimuli, which offer additional advantages to modulate the properties of the materials[7, 8]. The diversity of side-chain modification to polypeptides is vast, and plenty of functionalized α-amino acid N-carboxyanhydride (NCA) monomers have been prepared to obtain polypeptides with desired functional properties.
In addition to the direct polymerization from functionalized NCA monomers, post-polymerization modification strategy has been developed as an efficient approach to expand the diversity of side-chain functionalities of polypeptides. The living/controlled ring opening polymerization (ROP) of the synthetic functional NCA monomers with clickable substitutes greatly promotes the preparation of stimuli-responsive polypeptides. In particular, the polypeptides containing alkyne/alkene groups allow for the facile and efficient modification using thiol-yne/-ene photo addition, which offers significant advantages for biomedical applications[9-11]. Moreover, the thioether linkages exhibit the redox-responsive behavior that often results in the reversible polarity/conformation transitions of the polypeptides[7, 8]. Deming and coworkers have reported that the partial oxidation of poly (l-methionine) could trigger the reversible helix-to-coil transitions[7]. They further demonstrated that methionine-containing polypeptides could undergo a wide variety of chemical alkylation, and some of them were completely reversible[12, 13]. We have previously reported a series of thermal-responsive OEGylated poly (l-cysteine) s via thiol-ene Michael addition[14]. The stimuli-responsive property of the self-assemblies was systematically investigated[15]. We lately synthesized a new family of thermal-and redox-responsive poly (l-glutamate) bearing Y-shaped OEG side-chains[16].
In this paper, we prepared a series of OEGylated thioether bond-containing polypeptides derived from poly (l-glutamate) s via a combination of ROP and click chemistry (Scheme 1). This design simultaneously incorporated thermal-responsive OEG units and redox-responsive thioether bonds by a one-step thiol-ene photo addition. Unlike our previous study[16], the thermal-responsive OEG units are linearly grafted onto each side-chain, resulting in different molecular architecture of the side chains. The influence of both OEG units and thioether linkages on LCST behavior was systematically investigated.
Figure Scheme 1.
The synthetic route to PALGn-g-EGx by a combination of ROP of ALG-NCA with thiol-ene photo addition
EXPERIMENTAL
Synthesis of Methoxy Oligo (ethylene glycol) Thiol Modified PALG by Thiol-ene Photochemistry (PALGn-g-EGx)
Methoxy oligo (ethylene glycol) thiols (OEGx-SH, x=2, 3, 4) were synthesized using oligo (ethylene glycol) monomethyl ethers according to the literature procedures[22]. A typical example is given for the synthesis of PALGn-g-EG3, in which the subscript n represents the average degree of polymerization (DP) for PALG. PALG125 (0.20 g, 1.20 mmol of alkenes), OEG3-SH (1.09 g, 6 mmol of thiols) and DMPA (31 mg, 0.12 mmol) were mixed in 4 mL of DMF in a 20 mL vial containing a stirring bar. The reaction mixture was purged using N2 for 20 min before irradiated with UV light. The reaction was performed at room temperature for 3 h. Then the reaction mixture was precipitated into large excess diethyl ether, and the procedure was repeated three times using methanol/diethyl ether mixture. The obtained product was further washed twice with diethyl ether and dried overnight under vacuum to give the product as a light yellow solid with 86% yield.
Characterization
1H-NMR spectra and 13C-NMR spectra were recorded on a Bruker AV400 FT-NMR spectrometer. The infrared spectroscopy measurement was performed using a Nicolet Avatar 330 FTIR spectrometer. The solid samples were milled with potassium bromide (Aldrich) at a mass ratio of 1:100, and pressed into a disk before IR measurement. Tandem gel permeation chromatography/laser light scattering (GPC/LLS) was performed at 50 ℃ using an SSI pump connected to Wyatt Optilab DSP and Wyatt DAWN EOS light scattering detectors with 0.02 mol/L LiBr in DMF as the eluent at a flow rate of 1.0 mL/min. All the samples for GPC/LLS were prepared at the concentrations of 5-10 mg/mL. The clouding points (CPs) were measured by monitoring the transmittance of a 500 nm light beam through a quartz sample cell at a concentration of 2 mg/mL on a Shimadzu UV-Vis spectrometer. The solution was heated and cooled at a rate of 0.5 K/min. The CP was defined as the temperature corresponding to 50% transmittance of aqueous solution during the heating process. Circular dichroism (CD) spectra were recorded on a JASCO J-815 CD spectropolarimeter. The solution was placed into a quartz cell with a path length of 1.0 mm with temperature controlled by a water bath, and the concentration of samples was 0.2 mg/mL. Ellipticity ([θ] in deg·cm2·dmol-1)=(millidegrees × mean residue weight) / (path length in mm × concentration of polypeptide in mg/mL). The α-helix contents of the polypeptides were calculated using the following equation: % α-helix=(− [θ222] + 3000) / 39000[17]. The UV irradiation experiments were carried out using a mercury vapor lamp (254-577 nm, λmax=365 nm, 500 W) as the UV-light source.
General Procedure for Oxidation of PALGn-g-EGx to PALGn-g-OEGx
The oxidization experiments followed the modified procedures as reported in the literarure[8, 13]. A typical example is given for the synthesis of PALG125-g-OEG3. PALG125-g-EG3 (20 mg, 0.057 mmol of thiolether groups) was dissolved in a mixed aqueous solution of 30% H2O2 (29 μL, 0.29 mmol), AcOH (10 μL) and DI water (961 μL) and heated to 38 ℃ for 16 h under stirring. Excess peroxide was quenched by 1 mol/L sodium thiosulfate in water (0.29 mL). The solution mixture was transferred to a 3500 MWCO dialysis bag, and dialyzed against Millipore water for 3 days with water changing twice per day. Dialyzed polymers were lyophilized to yield partially oxidized products, PALG125-g-OEG3 (93% yield).
Synthesis of Poly (γ-allyl-l-glutamate) (PALG)
PALG was obtained by ROP of ALG-NCA using HMDS as the initiator (Scheme 1). All the polymerizations were performed in a high-purity N2-filled glovebox. Typically, a solution of HMDS (0.1 mol/L in DMF) was added to a solution of ALG-NCA (50 mg/mL in THF/DMF, 1/1, V/V). The solution was stirred at room temperature, and the polymerization progress was monitored by FTIR. The final solutions were precipitated into diethyl ether. Solids were collected by centrifugation, which was further purified by redissolved in CH2Cl2, followed by precipitated from diethyl ether. The products were dried under reduced pressure to yield white sticky solids.
General Procedure for Reduction of PALGn-g-OEGx
The reduction experiments followed the reported procedure[7]. PALGn-g-OEGx (x=3 and 4) at 10 mg/mL was dissolved in DI water containing thioglycolic acid (10 eq. of sulfoxide groups). The mixture was stirred at room temperature overnight and then diluted with equal amount water before transferring to a 3500 MWCO dialysis tubing. The solution mixture was dialyzed against Millipore water for 3 days with water changing twice per day. Dialyzed polypeptides were lyophilized to yield white solids, PALGn-g-*EGx (90%-93% yield).
Materials
Hexane, tetrahydrofuran (THF), and dichloromethane (DCM) were deoxygenated and dried by purging with nitrogen and passaging through activated alumina columns prior to use. Ethyl acetate (EtOAc) was freshly distilled from CaH2. Anhydrous N, N-dimethylformamide (DMF) was purchased from Alfa Aesar and used under an inert and dry atmosphere. Deionized water (DI water, 18 MΩ·cm) was obtained from a Millipore Milli-Q purification unit. l-Glutamic acid was purchased from GL Biochem (Shanghai) Ltd. Hexamethyldisilazane (HMDS) was purchased from Sigma-Aldrich. 2, 2-Dimethoxy-2-phenylacetophenone (DMPA) was purchased from Aladdin reagent. All other chemicals were purchased from commercial suppliers and used without further purification unless otherwise noted.
Synthesis of γ-Allyl-l-glutamate N-carboxyanhydride (ALG-NCA)
γ-Allyl-l-glutamate (ALG) was prepared by direct coupling of allyl alcohol and l-glutamic acid via sulfuric acid catalyzed esterification[11, 18-20]. ALG was purified according to previous work[21]. After purification, the ALG monomer (5.0 g, 27 mmol) was converted into corresponding NCA using triphosgene (2.9 g, 9.8 mmol) in THF (100 mL) at 50 ℃ under nitrogen. The solvent was removed under reduced pressure to give slightly brown oil, which was subsequently purified by the flash column chromatography method (note: the silica gel was dried at 120 ℃ over 6 h before use) with hexane as the eluent containing EtOAc gradient (from 30% to 50%). The organic solvent was then evaporated to give ALG-NCA as light yellow oil with 65% yield.
2.7 General Procedure for Oxidation of PALGn-g-EGx to PALGn-g-O2EGx
A typical example was given for the synthesis of PALG125-g-O2EG3. PALG125-g-EG3 (20 mg, 0.057 mmol of thiolether groups) was dissolved in a mixed aqueous solution of 30% H2O2 (57 μL, 0.57 mmol) and AcOH (1 mL). The mixture was stirred at room temperature for 16 h (about 20 ℃) under stirring. Excess peroxide was quenched by 1 mol/L sodium thiosulfate in water (0.57 mL). The solution mixture was transferred to a 3500 MWCO dialysis tubing and dialyzed against Millipore water for 3 days with water changes twice per day. Dialyzed polypeptides were lyophilized to yield completely oxidative products, PALG125-g-O2EG3 (91% yield).
RESULTS AND DISCUSSION
Redox-responsive Behaviors
It is known that thioethers can be selectively oxidized into polar sulfoxide or sulfone groups[8, 14] and the partial oxidation is reversible (Scheme 2)[7]. To study the redox property of PALGn-g-EGx, we performed different levels of chemical oxidation and reduction. H2O2 was first chosen as a model oxidant, and the partially oxidized sulfoxide and fully oxidized sulfone polymers were denoted as PALGn-g-OEGx and PALGn-g-O2EGx, respectively. 1H-NMR measurement and FTIR spectroscopy were used for examining their corresponding structures (Figs. 5, 6a and S8-S10). Figure 5(a) shows a couple of broad peaks between δ=2.45 and 2.80, which can be associated to methylene groups (d and e) (-CH2CH2SCH2CH2-) in PALGn-g-EG3. For the partially oxidized PALGn-g-OEG3, the pristine peaks at δ=2.45-2.80 completely disappear and the new peaks at d=2.86-3.37 appear (Fig. 5b), indicating the formation of sulfoxides. As further oxidized to sulfones (PALGn-g-O2EG3), a larger downfield shift of methylene groups (d and e in Fig. 5c) are observed, which can presumably be attributed to the methylene groups in the β position of sulfones (-CH2CH2S (O2) CH2CH2-). FTIR data show an increase of infrared absorption band at 1030 cm-1 (nS=O band, Fig. 6a) for PALGn-g-OEGx and two types of new FTIR bands at 1291 and 1321 cm-1 (symmetric and asymmetric stretching of -SO2-groups) for PALGn-g-O2EGx, consistent with the 1H-NMR results. To explore the reduction property of the samples, thioglycolic acid was used as a reductant, and the reducing products were denoted as PALGn-g-*EGx. The resonances of methylene groups at δ=2.45−2.80 and 2.86-3.37 indicate the coexistence of thioether and sulfoxide groups in PALGn-g-*EGx (Figs. 5d and S9d). We thus increase the amount of reducing reagent to 10 eq. with respect to sulfoxide groups. A decrease of infrared absorption band at 1030 cm-1 was clearly observed in PALGn-g-*EGx, indicating the successful reduction. GPC traces suggest that the PALG backbones and ester groups on the side-chains are barely affected (e.g.peak slightly broadening) during the oxidation and reduction process (Fig. S11).
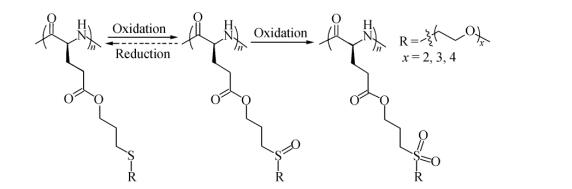 Figure Scheme 2.
Structures and redox properties of PALGn-g-EGx homopolypeptides
Figure Scheme 2.
Structures and redox properties of PALGn-g-EGx homopolypeptides
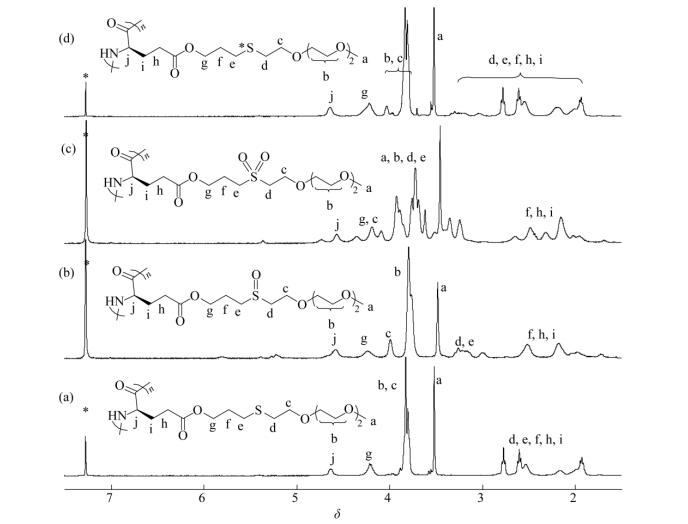 Figure 5.
1H-NMR spectra of (a) PALGn-g-EG3; (b) PALGn-g-OEG3; (c) PALGn-g-O2EG3 and (d) PALGn-g-*EG3 in CDCl3/CF3CO2D (85/15, V/V)
Figure 5.
1H-NMR spectra of (a) PALGn-g-EG3; (b) PALGn-g-OEG3; (c) PALGn-g-O2EG3 and (d) PALGn-g-*EG3 in CDCl3/CF3CO2D (85/15, V/V)
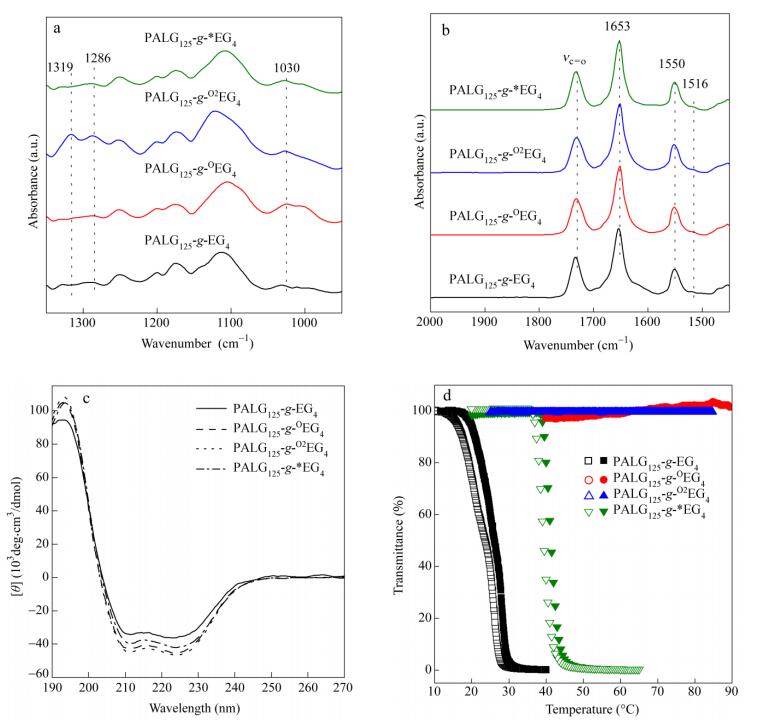 Figure 6.
(a) and (b) FTIR spectra in the solid state with the ranges of 1350−950 cm-1 and 2000-1450 cm-1, (c) CD and (d) variable-temperature UV spectra (solid symbol: heating; open symbol: cooling) of PALG125-g-EG4, PALG125-g-OEG4, PALG125-g-O2EG4 and PALG125-g-*EG4
Figure 6.
(a) and (b) FTIR spectra in the solid state with the ranges of 1350−950 cm-1 and 2000-1450 cm-1, (c) CD and (d) variable-temperature UV spectra (solid symbol: heating; open symbol: cooling) of PALG125-g-EG4, PALG125-g-OEG4, PALG125-g-O2EG4 and PALG125-g-*EG4
We further investigated the effects of redox properties on the secondary structures of PALGn-g-EGx using FTIR and CD spectroscopy. The secondary structure of PALG125-g-EG4 generally remains during the oxidation and subsequent reduction process, as indicated by strong absorbance at 1653 (amide I) and 1550 cm-1 (amide II) (Figs. 6b and S10b). Furthermore, similar CD spectra of PALG125-g-EG4 with nearly 100% helicity are observed in all cases (Figs. 6c and S12), indicating that the helical conformation is quite stable during the redox process. Interestingly, previously reported PPLG130-g-O2EG4 shows lower helical content than that of PALG125-g-O2EG4[16], probably because the linear PALGn-g-EGx side-chains with less fraction of thioether groups are not sufficient to destabilize the helicity of polypeptide backbones.
The molecular polarity of thioether groups can be largely increased upon oxidation. The increased polarity can further affect the thermal-responsive ability of the polymers due to the disruption of pristine hydrophilic/hydrophobic balance[7, 8, 13]. We thus studied the influence of redox properties on thermal-responsiveness of PALGn-g-EGx. As expected, increased solubility and CPs were observed for the sulfoxide and sulfone derivatives. PALG125-g-EG2, insoluble in water at room temperature, displays good solubility and reversible LCST behavior with the CP of 30 ℃ after partially oxidation with the degree of oxidation of~30% (Fig. S13a). Fully conversion to PALG125-g-O2EG2 results in completely water-soluble products without showing observable thermal-responsive behavior up to 85 ℃. Similarly, all sulfoxide and sulfone derivatives of polypeptides with x=3 and 4 show good water solubility with the absence of LCST behavior (Figs. 6d, S13b and S14). Figure 6(d) shows that PALG125-g-*EG4 regains LCST after reduction by thioglycolic acid. The CP of PALG125-g-*EG4 is determined to be 41 ℃, an intermediate value between the CP of PALG125-g-EG4 (26.3 ℃) and the CP of PALG125-g-OEG4 (above 85 ℃), suggesting partial reduction of sulfoxide groups[7]. The degree of reduction was calculated to be 80% by 1H-NMR. Similarly, all the other reducing products (PALG125-g-*EG3, PALG68-g-*EG3 and PALG68-g-*EG4) show reversible LCST-type phase transition (Figs. S13 and S14), indicating that the redox-reaction offers an efficient way to finely tune the water solubility and thermal-responsive property of the materials.
Secondary Structure
It is generally accepted that the inherent secondary structures of polypeptides are dependent on both main chain lengths and side-chain structures[19, 26]. FTIR and CD spectroscopy were used to investigate the secondary structures of the polypeptides in solid (Figs. 2b and S3) and in solution (Figs. 2c and S4), respectively. PALG68 and PALG125 show strong absorption bands centered at 1653 (amide I) and 1550 cm-1 (amide II), indicative of the dominant a-helix structure (Fig. S3a)[27]. Notably, there are a broad band about 1630-1660 cm-1 and a weak band around 1516 cm-1, which suggest the coexistence of b-sheet or random coil[28]. In comparison to PALG68, PALG125 shows much stronger band intensity at 1653 and 1550 cm-1 and decreased absorbance at 1516 cm-1, indicative of enhanced a-helicity. In the case of PALGn-g-EGx, the sharp absorption bands at 1653 (amide I) and 1550 cm-1 (amide II) and a trivial absorption at 1516 cm-1 suggest that the OEG-functionalized samples mainly adopt the a-helical structure (Figs. 2b and S3b)[21]. The CD spectra of PALGn-g-EG4 in aqueous solutions confirm the helix conformation with a positive peak close to 195 nm and two negative peaks at 208 and 222 nm. Note that the PALGn-g-EG3 samples are insoluble in water at room temperature, the CD measurement are thus performed in a mixture of H2O/EtOH (1/1, V/V). A typical helix conformation is observed as shown in Fig. S4. We therefore demonstrate that the helical structure of the polypeptides can be stabilized by OEGx side-chains[21].
Synthesis of PALGn-g-EGx
As illustrated in Scheme 1, PALG was prepared by the ROP of an alkene-substituted l-glutamate NCA (ALG-NCA) using HMDS as an initiator in a mixture of THF/DMF (1/1, V/V)[23]. The chemical structures of ALG-NCA and the corresponding PALG were determined by 1H-NMR, and all the proton signals were well assigned (Figs. 1a and 1b). The number-average molecular weight (Mn) and dispersity (
) of PALG were further determined by GPC/LLS (Figs. 2a and S2, solid line). By varying the molar ratio of the NCA monomer to initiator, we synthesized the polymers with two different DP of 68 and 125, denoted as PALG68 and PALG125, respectively. Both PALG68 and PALG125 showed relatively narrow molecular weight distribution with < 1.2, summarized in Table 1. Note that the DP of the samples are designed nearly same as the previously reported poly (γ-propargyl-l-glutamate) s, e.g. PPLG65 and PPLG130, which enables direct comparison of both polypeptides[16]. 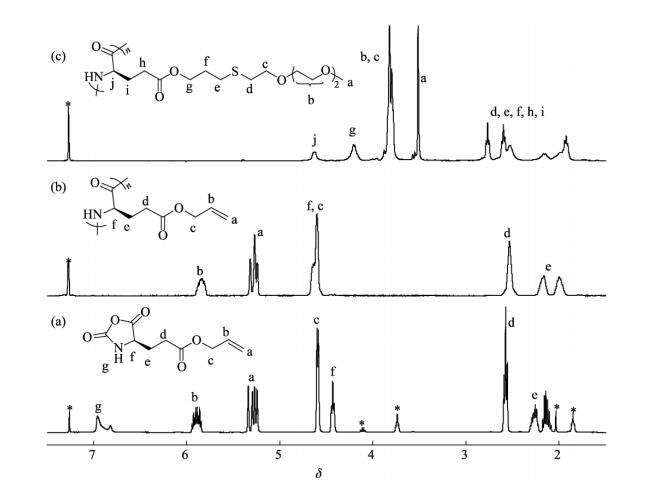 Figure 1.
1H-NMR spectra of (a) ALG-NCA in CDCl3 (* indicates CDCl3, THF and EtOAc residue), (b) PALG and (c) PALGn-g-EG3in CDCl3/CF3CO2D (85/15, V/V)
Figure 1.
1H-NMR spectra of (a) ALG-NCA in CDCl3 (* indicates CDCl3, THF and EtOAc residue), (b) PALG and (c) PALGn-g-EG3in CDCl3/CF3CO2D (85/15, V/V)
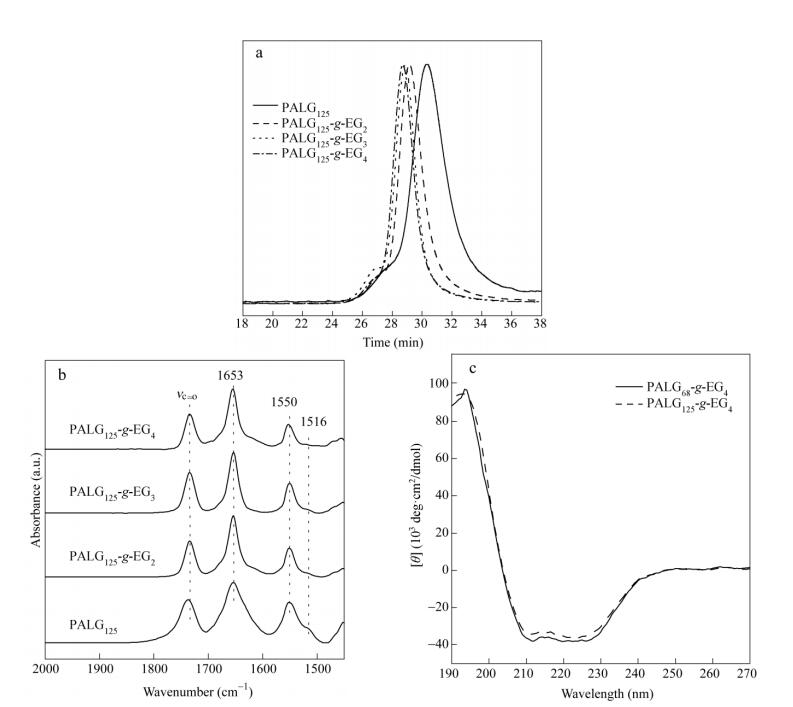 Figure 2.
GPC chromatographs (a) and FTIR spectra (b) of PALG125, PALG125-g-EG2, PALG125-g-EG3 and PALG125-g-EG4; (c) CD spectra of PALG68-g-EG4and PALG125-g-EG4 (0.2 mg/mL in DI water) at 20 ℃
Figure 2.
GPC chromatographs (a) and FTIR spectra (b) of PALG125, PALG125-g-EG2, PALG125-g-EG3 and PALG125-g-EG4; (c) CD spectra of PALG68-g-EG4and PALG125-g-EG4 (0.2 mg/mL in DI water) at 20 ℃
 Table 1.
Molecular parameters of PALG
Table 1.
Molecular parameters of PALG
Entry [M]/[I] a Mnb (kDa) b DP Yield (%) 1 100 11.4 1.13 68 81 2 150 21.0 1.17 125 84 a Molar ratio of ALG-NCA to HMDS; b Molecular weight and dispersity ( ) determined by GPC/LLS Table 1. Molecular parameters of PALGThree types of synthetic thiol-terminated oligo (ethylene glycol) monomethyl ethers, OEGx-SH (x=2-4), were grafted onto the PALG backbone via thiol-ene photo addition (Scheme 1). The chemical structures of OEGx grafted polypeptides (denoted as PALGn-g-EGx) were confirmed by 1H-NMR and GPC. The proton signals at δ=5.19-5.39 and 5.74-5.97 almost disappeared, and the new peaks at δ=3.42-3.58 (a) and δ=3.67-3.95 (b and c) were ascribed to the protons of OEG moiety, which suggested the successful conversion of allyl groups and the grafting of OEGx (Figs. 1c and S1). To obtain the samples with high grafting efficiency, the addition reactions were conducted with high concentrations of reactants in the presence of a photo initiator (∼5 wt% of PALG in DMF, molar ratio [allyl groups]:[OEGx-SH]=1:5). Conversion of the allyl groups could be improved to 92%-100%. The GPC traces of PALGn-g-EGx (Figs. 2a and S2) show mono-modal peaks at longer elution time than that of PALG homopolymer precursor, further confirming the successful conjugation of the OEG side-chains with negligible side reactions. All the PALGn-g-EGx samples have good solubility in common organic solvents. However, they behave very differently in water (Table S1). PALGn-g-EG2 and PALGn-g-EG3 are insoluble in water at ambient temperature while cooling the PALGn-g-EG3 solution to 0 ℃ results in full dissolution in water. Instead, PALGn-g-EG4 is soluble in water in all above mentioned conditions, indicative of the improved hydrophilicity due to the longer OEG side-chains[24, 25].
Thermal-responsive Behaviors
The effects of temperature on the solution properties of the samples PALGn-g-EG3 and PALGn-g-EG4 were then investigated by turbidity measurement. Corresponding CPs were determined at the temperature with 50% transmittance. In all cases, the transmittance of the solutions decreases upon heating, indicating an expected thermal-responsive behavior (Figs. 3a and S5a). Figure 3(a) displays plots of the transmittance versus temperature for PALG125-g-EG3 and PALG125-g-EG4 aqueous solutions at a concentration of 2 mg/mL. PALG125-g-EG3 shows characteristic LCST behaviour in the temperature range from 1 ℃ to 30 ℃ with the CP of 5.5 ℃. 66% transmittance is recovered during the cooling run. Further cooling the solution to 0 ℃ and keeping for 30 min results in the complete recovery of transmittance. This is probably because the CP is close to the freezing point and the rehydration of OEG units requires slight overcooling to overcome the energy barriers[29]. In comparison, PALG125-g-EG4, with one more OEG unit in each side-chain, shows increased solubility with a CP of 26.3 ℃ under the same conditions. Furthermore, a complete recovery of transmittance after 6 heating-cooling cycles between 15 and 40 ℃ is observed (Fig. 3b). The CP determined from the cooling ramp is about 3.5 and 2.7 ℃ lower than that from the heating ramp for PALG125-g-EG3 and PALG125-g-EG4, respectively. The hysteresis probably corresponds to the above-mentioned energy barriers in the rehydration of OEG units[29].
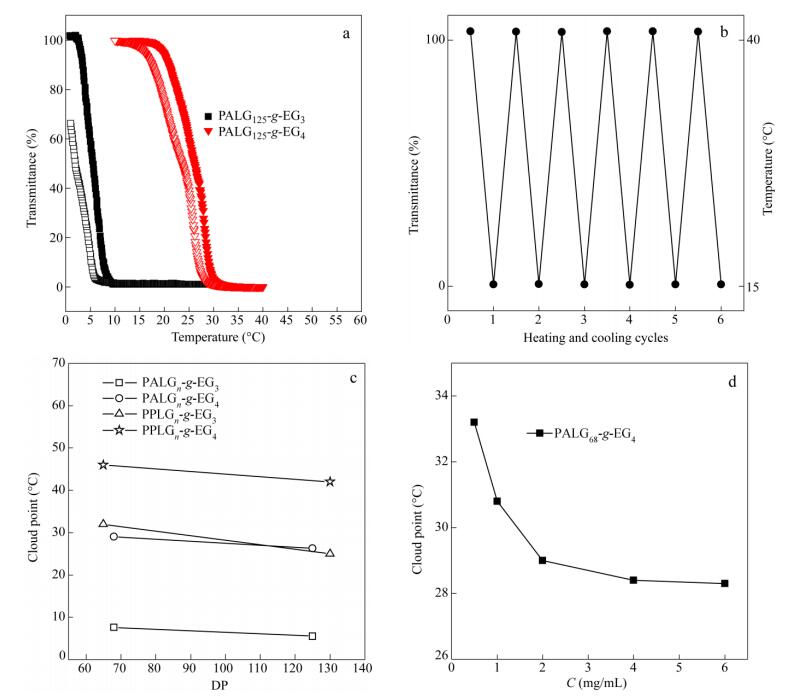 Figure 3.
(a) Plots of transmittance as a function of temperature for the samples (2 mg/mL in DI water) of PALG125-g-EG3 and PALG125-g-EG4 (solid symbol: heating; open symbol: cooling); (b) optical transmittance of aqueous PALG125-g-EG4 versus 6 heating-cooling cycles between 15 and 40 ℃, 10 min/cycle; (c) cloud point dependence on DP for PALGn-g-EGx and PPLGn-g-EGx (x=3 or 4) (all the samples were prepared at 2 mg/mL.); (d) cloud points of PALG68-g-EG4 measured in different polymer concentration
Figure 3.
(a) Plots of transmittance as a function of temperature for the samples (2 mg/mL in DI water) of PALG125-g-EG3 and PALG125-g-EG4 (solid symbol: heating; open symbol: cooling); (b) optical transmittance of aqueous PALG125-g-EG4 versus 6 heating-cooling cycles between 15 and 40 ℃, 10 min/cycle; (c) cloud point dependence on DP for PALGn-g-EGx and PPLGn-g-EGx (x=3 or 4) (all the samples were prepared at 2 mg/mL.); (d) cloud points of PALG68-g-EG4 measured in different polymer concentration
We then investigated the effect of DP on the CP for PALGn-g-EG3 and PALGn-g-EG4. With the same DP, PALGn-g-EG3 shows a lower CP than that of PALGn-g-EG4, suggesting the enhanced hydrophilicity of the sample with longer OEG side-chain length (Fig. 3c). As the OEG side-chain length is fixed, the CP decreases with increasing the DP of PALG backbones[30]. As the DP increases from 68 to 125, the CP of PALGn-g-EG3decreases from 7.6 ℃ to 5.5 ℃ and the CP of PALGn-g-EG4 decreases from 29 ℃ to 26.3 ℃. The phase transition is reversible for multiple cycles between 20 and 40 ℃ (Fig. S5b). To investigate the relationship between structure and property, we compared the CPs of PALGn-g-EGx with those of previously reported PPLGn-g-EGx, i.e.OEGxmodified PPLG by thiol-yne photochemistry[16]. Unlike the linear side-chain structure of PALGn-g-EGx, PPLGn-g-EGx has a Y-shaped molecular architecture with two thermal-responsive OEG units on each side-chain. With the same OEG side-chain length and similar DP, a higher CP is observed in Y-shape PPLGn-g-EGx (Figs. 3c and S6). For example, the CPs of PALG125-g-EG3 and PALG125-g-EG4 are 20 and 16 ℃, lower than those of PPLG130-g-EG3 and PPLG130-g-EG4, respectively. Similarly, the CPs of PALG68-g-EG3 and PALG68-g-EG4 are about 24 and 17 ℃, lower than those of PPLG65-g-EG3 and PPLG65-g-EG4, respectively. This is possibly due to the enhanced hydrophilicity of Y-shaped side-chain architecture. The influence of the polymer concentration on the CP was further investigated, shown in Fig. 3(d). It is evident that the CP of PALG68-g-EG4 decreases monotonically with increasing polymer concentration, possibly due to the enhanced intermolecular association strength in concentrated solution (Fig. S5c)[31]. As the polymer concentration further increases, the CP is less dependent on the concentration. It is demonstrated that the CP of the polymer can be finely tuned by changing the chain length, architecture of OEGx groups, the DP of PALG backbone and the polymer concentration.
The stability of the secondary structure as a function of temperature was measured by temperature-varied CD measurement. In a typical CD plot of PALG125-g-EG4 at different temperatures, the CD signals decrease significantly as the temperature is above the corresponding CP (Figs. 4a and S7a). The calculated a-helicity of the polypeptides declines as well (Table S2). The CD signals are recovered during the cooling scan (Figs. 4b and S7b). We therefore demonstrate that the variation of CD signals is due to the reversible aggregation of the molecules without disrupting the preformed secondary structure[14]. Similar results are also observed for PALG68-g-EG4, which are consistent with turbidity results.
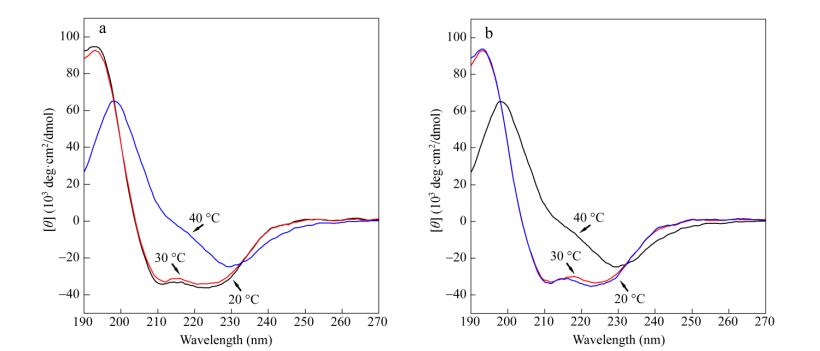 Figure 4.
CD spectra of PALG125-g-EG4 as a function of temperature: (a) heating scan and (b) cooling scan
Figure 4.
CD spectra of PALG125-g-EG4 as a function of temperature: (a) heating scan and (b) cooling scan
Conclusions
We demonstrated the preparation of thermal-and redox-responsive polypeptides that were composed of a poly (l-glutamate) backbone and the OEGx side-chains with thioether linkages via NCA ring-opening polymerization and thiol-ene photoaddition. The molecular structures and physical properties were characterized thoroughly. Depending on the chain length of OEGx side-chains, PALGn-g-EGx showed different water solubility. In particular, the samples with x=3 and 4 displayed a LCST-type phase transition upon heating. The cloud points were significantly affected by the OEGxlength, polypeptide molecular weights, and the sample concentrations, suggesting a highly tunable phase transition behavior. Additionally, oxidation/reduction as a second stimulus played a key role in tuning water solubility and the CPs without disrupting the secondary structures. Overall, this synthetic approach allows for the facile preparation of PALGn-g-EGx polymers with multi-responsive properties and reversible phase transition behavior, making them promising candidates for multi-stimuli responsive polymeric materials.
-
-
[1]
Stuart, M.A., Huck, W.T., Genzer, J., Muller, M., Ober, C., Stamm, M., Sukhorukov, G.B., Szleifer, I., Tsukruk, V.V., Urban, M., Winnik, F., Zauscher, S., Luzinov, I. and Minko, S., Nat. Mater., 2010, 9(2):101 doi: 10.1038/nmat2614
-
[2]
Hoffman, A.S., Adv. Drug Delivery Rev., 2013, 65(1):10 doi: 10.1016/j.addr.2012.11.004
-
[3]
Tian, C., Ling, J. and Shen, Y.Q., Chinese J. Polym. Sci., 2015, 33(8):1186 doi: 10.1007/s10118-015-1669-0
-
[4]
He, C., Zhuang, X., Tang, Z., Tian, H. and Chen, X., Adv. Healthcare Mater., 2012, 1(1):48 doi: 10.1002/adhm.201100008
-
[5]
Lu, H., Wang, J., Song, Z., Yin, L., Zhang, Y., Tang, H., Tu, C., Lin, Y. and Cheng, J., Chem. Commun., 2014, 50(2):139 doi: 10.1039/C3CC46317F
-
[6]
Shen, Y., Fu, X., Fu, W. and Li, Z., Chem. Soc. Rev., 2015, 44(3):612 doi: 10.1039/C4CS00271G
-
[7]
Kramer, J.R. and Deming, T.J., J. Am. Chem. Soc., 2014, 136(15):5547 doi: 10.1021/ja500372u
-
[8]
Kramer, J.R. and Deming, T.J., J. Am. Chem. Soc., 2012, 134(9):4112 doi: 10.1021/ja3007484
-
[9]
Brosnan, S.M. and Schlaad, H., Polymer, 2014, 55(22):5511 doi: 10.1016/j.polymer.2014.08.067
-
[10]
Sun, J. and Schlaad, H., Macromolecules, 2010, 43(10):4445 doi: 10.1021/ma100401m
-
[11]
Huang, Y., Zeng, Y., Yang, J., Zeng, Z., Zhu, F. and Chen, X., Chem. Commun., 2011, 47(26):7509 doi: 10.1039/c1cc12177d
-
[12]
Kramer, J.R. and Deming, T.J., Chem. Commun., 2013, 49(45):5144 doi: 10.1039/c3cc42214c
-
[13]
Gharakhanian, E.G. and Deming, T.J., J. Phys. Chem. B, 2016, 120(26):6096 doi: 10.1021/acs.jpcb.6b02065
-
[14]
Fu, X., Shen, Y., Fu, W. and Li, Z., Macromolecules, 2013, 46(10):3753 doi: 10.1021/ma400678w
-
[15]
Fu, X., Ma, Y., Shen, Y., Fu, W. and Li, Z., Biomacromolecules, 2014, 15(3):1055 doi: 10.1021/bm5000554
-
[16]
Fu, X., Ma, Y., Sun, J. and Li, Z., RSC Adv., 2016, 6(74):70243 doi: 10.1039/C6RA17427B
-
[17]
Morrow, J.A., Segall, M.L., Lund-Katz, S., Phillips, M.C., Knapp, M., Rupp, B. and Weisgraber, K.H., Biochemistry, 2000, 39(38):11657 doi: 10.1021/bi000099m
-
[18]
Xiao, C., Zhao, C., He, P., Tang, Z., Chen, X. and Jing, X., Macromol. Rapid Commun., 2010, 31(11):991 doi: 10.1002/marc.200900821
-
[19]
Tang, H.Y. and Zhang, D.H., Polym. Chem., 2011, 2(7):1542 doi: 10.1039/c1py00015b
-
[20]
Wu, L.Y., Yu, L., Fu, X.H. and Li, Z.B., Chininese J. Polym. Sci., 2015, 33(8):1140 doi: 10.1007/s10118-015-1640-0
-
[21]
Chen, C., Wang, Z. and Li, Z., Biomacromolecules, 2011, 12(8):2859 doi: 10.1021/bm200849m
-
[22]
Snow, A.W. and Foos, E.E., Synthesis-Stuttgart, 2003, (4):509
-
[23]
Lu, H. and Cheng, J., J. Am. Chem. Soc., 2007, 129(46):14114 doi: 10.1021/ja074961q
-
[24]
Hu, Z.B., Cai, T. and Chi, C.L., Soft Matter, 2010, 6(10):2115 doi: 10.1039/b921150k
-
[25]
Lutz, J.F., Adv. Mater., 2011, 23(19):2237 doi: 10.1002/adma.v23.19
-
[26]
Yuan, Q.L., Liu, W.J., Deng, Y., Ling, Y. and Tang, H.Y., Chinese J. Polym. Sci., 2015, 33(8):1150 doi: 10.1007/s10118-015-1665-4
-
[27]
Blout, E.R. and Asadourian, A., J. Am. Chem. Soc., 1956, 78(5):955 doi: 10.1021/ja01586a022
-
[28]
Haris, P.I. and Chapman, D., Biopolymers, 1995, 37(4):251 doi: 10.1002/(ISSN)1097-0282
-
[29]
Lutz, J.F., Weichenhan, K., Akdemir, Ö. and Hoth, A., Macromolecules, 2007, 40(7):2503 doi: 10.1021/ma062925q
-
[30]
Zhang, S.S., Chen, C.Y. and Li, Z.B., Chinese J. Polym. Sci., 2013, 31(2):201 doi: 10.1007/s10118-013-1218-7
-
[31]
Cheng, Y.L., He, C.L., Xiao, C.S., Ding, J.X., Zhuang, X.L. and Chen, X.S., Polym. Chem., 2011, 2(11):2627 doi: 10.1039/c1py00281c
-
[1]
-

 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 778
- HTML全文浏览量: 19
 下载:
下载:
