 图14
Schematic representation of the silver nanoparticles formation in the crosslinked-chitosan network
Figure14.
Schematic representation of the silver nanoparticles formation in the crosslinked-chitosan network
图14
Schematic representation of the silver nanoparticles formation in the crosslinked-chitosan network
Figure14.
Schematic representation of the silver nanoparticles formation in the crosslinked-chitosan network
Citation:
Hossein Ghasemzadeh, Mahdieh Sheikhahmadi, Fatemeh Nasrollah. Full Polysaccharide Crosslinked-chitosan and Silver Nano Composites, for Use as an Antibacterial Membrane[J]. Chinese Journal of Polymer Science,
2016, 34(8): 949-964.
doi:
10.1007/s10118-016-1807-3

Full Polysaccharide Crosslinked-chitosan and Silver Nano Composites, for Use as an Antibacterial Membrane
English
Full Polysaccharide Crosslinked-chitosan and Silver Nano Composites, for Use as an Antibacterial Membrane
Abstract:
Full polysaccharide crosslinked-chitosan membranes were prepared by crosslinking of chitosan with chitosan dialdehyde followed by reduction with sodium borohydride. Partially oxidized chitosan, generated from periodate oxidation of chitosan, was used as a crosslinker. The modulus values and elongation at break were increased with increasing the crosslinker weight ratio. The rheological measurements showed that depolymerization of chitosan can take place rapidly in the presence of the oxidizing agent. The weight reduction of crosslinked-chitosan membrane after 12 h, at pH=4 and pH=2 was found to be 85.0% and 90.0%, respectively. The structure of the crosslinked-chitosan and the silver nanocomposite were confirmed by FTIR spectroscopy, scanning electron microscopy (SEM), and thermogravimetric analysis (TGA). Transmission electron microscopy (TEM) reveals the presence of well-separated Ag nanoparticles with diameters in the range of 4-10 nm. The silver ion loading increases with increasing the silver ion concentration, and decreasing the crosslink density. The MBC/MIC ratio of 2.0, 2.0, and 1.0 was achieved for E. coli, S. aureus, and P. aeruginosa, respectively.
-
Key words:
- Silver nanocomposites
- / Chitosan
- / Crosslinking
- / Antibacterial effects
-
INTRODUCTION
Biodegradable and biocompatible materials have wide applications in the field of medicine, pharmaceutics, tissue engineering, and metal chelation[1-4]. From the point of view of both synthetic and mechanistic interest, much attention has been paid to developing bio-degradable materials from a sustainable resource. In particular, polysaccharides have been used for production of nanoparticles as a green synthesis process. Polysaccharides have also great potential applications in biotechnology and drug delivery systems[5-8]. Chitosan has been described as a non-toxic, biodegradable and biocompatible polymer with very interesting biological properties, such as mucoadhesive, anticoagulant and antimicrobial activity. It has also been used in several applications such as biomedical, pharmaceutical and biotechnological fields as well as in the food industry[9-12]. Chitosan is sensitive to a variety of degradation including oxidative-reductive free radical depolymerization as well as enzyme, acid and base catalyzed degradation[13]. Several crosslinking agents such as epichlorohydrin[14, 15], formaldehyde[16, 17], glutaraldehyde[18, 19], and ethylene glycol diglycidyl ether[20, 21], have been employed to crosslink the chitosan polysaccharide. However, the toxicity of these chemicals limits their utility and the biocompatibility of the products. To overcome this inconvenience, use of natural crosslinking agents is a superior approach to achieve full natural-based network for biomaterial applications. Genipin, has been used as a natural crosslinker for crosslinking of the chitosan[22-25]. Dialdehyde derivatives prepared from periodate oxidation of some polysaccarides such as starch[26], cellulose[27], β-cyclodextrin[28], dextran[29], carboxymethyl cellulose[30], and xanthan gum[31], have been also employed to crosslinking of chitosan, collagen and gelatin. In this work partially oxidized chitosan, generated from periodate oxidation of chitosan, was used as a cross-linker to form a macromolecular network. The effects of crosslinker concentration, pH, and salt solutions on the swelling behavior of the product were studied. It was found that the stress-strain relations and mechanical properties of the prepared films were improved by crosslinking. The silver nanocomposite was prepared via in situ reduction of silver nitrate (AgNO3) using sodium borohydride (NaBH4) as a reducing agent. The structure of the synthesized network and the silver nanocomposites was confirmed by solubility test, FTIR spectroscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and thermogravimetric analysis (TGA). The silver ion loading increases with increasing the silver ion concentration, and decreasing the crosslinke density. The silver nanocomposites exhibit an inhibition zone against both Gram-positive and Gram-negative bacteria[32, 33]. The MBC/MIC ratio of 2.0, 2.0, and 1.0 was achieved for E. coli, S. aureus, and P. aeruginosa, respectively.
EXPERIMENTAL
Microscopic Analysis
The surface morphology was analyzed by scanning electron microscopy, SEM (Philips, XL30) operated at 25 kV. The samples were coated with a thin layer of gold metal by plasma vapor deposition. A Zeiss-EM10C transmission electron microscope (TEM) operating at 80 kV was employed for TEM studies. The samples were homogenized by ultrasonic homogenizer in water for 30 min. Then, the samples were prepared on a 300 mesh carbon-coated copper grid.
FTIR Analysis
The Fourier transform infrared spectra (FTIR) of thin film samples were obtained using a Bruker Tensor 27 FTIR spectrophotometer.
Preparation of the Silver Nanocomposites
To prepare silver nanocomposites, accurately weighed dry samples were equilibrated with water for 3 days at room temperature. Then, the samples were transferred to another beaker containing 50 mL of 5 mmol/L AgNO3 aqueous solution and allowed to equilibrate for 2 days. In this step, the silver ions are exchanged from solution into the networks by binding with―NH2, and―OH groups of the chitosan chains and the rest of metal ions are occupied in free-network spaces of the crosslinked-chitosan. After washing with distilled water, the silver salt loaded samples were transferred into a beaker containing 50 mL of 25 mmol/L NaBH4 aqueous solution and allowed for 3 h to reduce the silver ions into silver nanoparticles. Subsequently, the samples were washed with distilled water and were dried at room temperature.
TGA Analysis
Thermal analysis was performed using a thermogravimetric analyzer (Perkin Elmer Pyris Diamond). The thermal analyzer was used for thermogravimetric analysis (TGA) of the samples under nitrogen atmosphere and the heating rate of 20 K/min.
Atomic Absorption Analysis
The amount of the silver ion loaded in the sample matrix was determined by a Varian model 220Z spectrophotometer. The influence of the initial concentration of the crosslinker, and silver nitrate on the extent of silver ion loading was studied by this method.
Preparation of the Oxidized Chitosan
Oxidation of chitosan was performed according to the procedure described by Vold et al[35]. Briefly, chitosan (0.20 g) was dissolved in 20.0 mL of distilled water by prolonged magnetic stirring in a beaker. Sodium metaperiodate (0.10 g, dissolved in 10 mL of distilled water) was added to the solution. The solution was stirred magnetically in a dark medium at 25 ℃ for 15 h. Then, the oxidized chitosan was purified by precipitation with the addition of NaOH (1.0 mol/L). The products was dissolved in water (20.0 mL) and re-precipitated by the addition of NaOH (1.0 mol/L). The process was repeated and then the precipitate was washed with ethanol.
Antibacterial Activity
The antibacterial activity of the silver nanocomposites was performed on Gram-positive and Gram-negative microorganisms. Agar medium was obtained from Merck. After dissolution in distilled water and sterilization, the agar medium was incubated for 24 h at 37 ℃. The disc diffusion method was performed on Mueller-Hinton agar and the zone of inhibition was measured using E. coli (ATCC, 10536), Pseudomonas aeruginosa (ATCC, 15442), and Staphylococcus aureus (ATCC, 6538) in nutrient agar medium, after 24 h of incubation. The experiment was repeated three times to ensure reproducibility. The minimum inhibitory concentration (MIC) and the minimum bactericidal concentration (MBC) were determined using E. coli (ATCC, 8739), Pseudomonas aeruginosa (ATCC, 9027), and Staphylococcus aureus (ATCC, 6538) in nutrient agar medium. The MIC was read after 24 h of incubation. The MBC was examined by inoculation of the samples to agar medium without antimicrobial agents and was determined as the lowest concentration that inhibits the visible growth of the used bacterium.
Rheological Measurements
Rheological properties of the chitosan solutions were measured at room temperature using a MCR 300 (Anton Paar, Austria) modular compact rheometer.
Swelling and Deswelling Kinetics
To study the rate of the swelling, certain amount of the samples (0.2±0.01 g) with the average particle mesh sizes between 40 and 60 (250-400 μm) was placed into a weighed tea bag and immersed in 200 mL of distilled water. At sequential time intervals, the swelling was measured according to the earlier mentioned method.
To investigate the rate of deswelling, a weighed tea bag containing determined amount of completely swellen samples was hung up to remove the excess water. The deswelling water ratio of the each sample was achieved using the following equation:
where Wt0 and Wt are the initial weight of the fully swollen sample and the weight of sample at the deswelling time t, respectively.
Mechanical Tests
The stress-strain test of crosslinked chitosan membranes in dry state was done using an Universal Testing Machine model STM-20, at room temperature with a cross-head speed 1 mm/min. All films were cut into 2.5 cm × 7 cm samples, whereas the film thickness was measured using a digital micrometer. The results were obtained from the mean values of 3 independent measurements. The Young’s modulus was calculated from the slope of the linear part of the stress-strain curve.
1H-NMR Analysis
1H-NMR spectra were recorded with a Bruker Avance, 300 MHz spectrometer in D2O and 1% TFA at room temperature.
Elemental Analysis
Elemental analysis was performed by a Perkin-Elmer 2004 (Ⅱ) CHN analyzer.
Swelling Test
Dry samples of the same weight were equilibrated in distilled water at room temperature for 24 h. The equilibrium swelling capacity was calculated using the following equation:
where Ws and Wd are the weights of the swollen and the dry samples, respectively. Accordingly, equilibrium swelling was determined as grams of water per gram of the sample (g/g).
Determination of Silver Ion Loading
The accurately weighed dried samples were dispersed in 1 wt% aqueous acetic acid solution to swell completely. The samples were washed several times with distilled water and then were equilibrated with distilled water for 24 h. Then, the completely swollen samples were transferred to a beaker containing 100 mL of AgNO3 aqueous solutions, with various concentrations for 24 h. Then the samples were filtered and the concentration of silver ion in the remaining solution was determined by atomic absorption spectroscopy.
Preparation of the Crosslinked-chitosan Membranes
For preparation of the crosslinked-chitosan membranes various amounts of oxidized chitosan were combined with determined amount of chitosan to prepare different weight ratios of the samples. Mixtures were cast into a plastic Petri plates (38.5 cm2). They were subsequently allowed to dry in an oven at 45 ℃ for 12 h. In this step, the conjugation between reactive aldehyde groups and primary amino groups of chitosan can take place. The membranes were washed with distilled water and then incubated in aqueous sodium borohydride (NaBH4) (0.5%) solution for 3 h, to reduce non-crosslinked aldehyde groups and to stabilize the product. The products were washed several times with distilled water, and were allowed to dry at room temperature.
Materials
Chitosan (Mr ≈ 1.5 × 105) with 55% degree of deacetylation (determined by CHN analysis) was obtained from Novin Shimyar Chemical Co. Sodium meta periodate was obtained from AppliChem Company. Both were used without further purification. Silver nitrate (AgNO3), and sodium borohydride (NaBH4) were purchased from Merck chemical Co. All the other reagents were of analytical grade.
Sol Content Determination
The soluble (sol) content was determined according to our previous work[34]. Briefly, the primitive crude products were dispersed in distilled water for 3 days to swell completely. The sample was washed with distilled water frequently and then filtered. Then it was kept in excess ethanol for 48 h, and dried at 50 ℃ for 12 h. The soluble content was calculated as the weight loss of the initial crude product.
RESULTS AND DISCUSSION
Synthesis of the Silver Nanocomposite
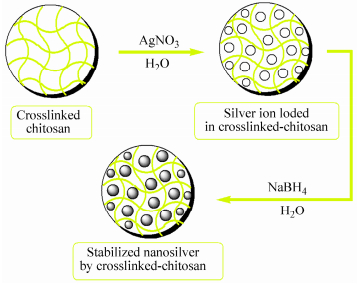
The crosslinked-chitosan network can act as a nanoreactor for stabilizing the silver nanoparticles. Preparation of silver nanoparticles by green synthesis approaches has advantages over conventional methods. In green synthesis process silver nanoparticles are prepared using water as an environmentally benign solvent and polysaccharides as capping agents[47], or in some cases polysaccharides serve as both a reducing and a capping agent[48]. For preparation of silver nanocomposite, the completely swollen pre-weighed crosslinked-chitosan membranes were loaded with the 5 mmol/L AgNO3 solution and then reduced in aqueous solution of NaBH4(Fig. 14). It is hypothesized that the silver nanoparticles are immobilized throughout the chitosan networks due to strong localization of the Ag+ ions within the network. This is caused by the complexation of the Ag+ ions by both the nitrogen atoms of chitosan and the oxygen atoms of the polysaccharide. The prepared silver nanoparticles in the chitosan networks are highly stable and did not show any signs of aggregation, even after storage for several months. Similar behavior was reported[49, 50].
图14
Schematic representation of the silver nanoparticles formation in the crosslinked-chitosan network
Figure14.
Schematic representation of the silver nanoparticles formation in the crosslinked-chitosan network
Figure 15 shows SEM photographs of the dry samples of crosslinked-chitosan with various amount of crosslinking agent, illustrating the surface with pores that are irregular in size and shape. It seems that the morphology of the membranes is affected by the amount of crosslinker at the same conditions of drying. Scanning electron microscopy image of the silver nanocomposite demonstrates fine and well-dispersed nanoparticles in the crosslinked-chitosan network (Fig. 15d). It is reported that the control or alignment of nanoparticles can be achieved by modifying the network architectures[51].
 图15
SEM photographs of the crosslinked-chitosan membranes (a) with 10% crosslinker, (b) with 30% crosslinker, (c) with 50% crosslinker, and (d) silver nanocomposite
Figure15.
SEM photographs of the crosslinked-chitosan membranes (a) with 10% crosslinker, (b) with 30% crosslinker, (c) with 50% crosslinker, and (d) silver nanocomposite
图15
SEM photographs of the crosslinked-chitosan membranes (a) with 10% crosslinker, (b) with 30% crosslinker, (c) with 50% crosslinker, and (d) silver nanocomposite
Figure15.
SEM photographs of the crosslinked-chitosan membranes (a) with 10% crosslinker, (b) with 30% crosslinker, (c) with 50% crosslinker, and (d) silver nanocomposite
Transmission electron microscopy images of the silver nanocomposite depicted in Fig. 16 indicate that silver nanoparticles are distributed in the optimized chitosan networks. The nanoparticles in the chitosan networks are stabilized by the presence of the chitosan in the networks. TEM micrographs reveal the presence of well-separated Ag nanoparticles with diameters in the range of 4-10 nm, as shown in Fig. 16.
 图16
TEM micrographs of silver nanocomposite dispersed in water with different magnifications
Figure16.
TEM micrographs of silver nanocomposite dispersed in water with different magnifications
图16
TEM micrographs of silver nanocomposite dispersed in water with different magnifications
Figure16.
TEM micrographs of silver nanocomposite dispersed in water with different magnifications
Silver Ion Loading in the Membrane Matrix
The effect of silver ion concentration and crosslinker weight ratio on the loading was studied. It is obtained that the silver ion loading increases with increasing the silver ion concentration, as shown in Table 2. However, the loading percent shows a decreasing trend.
 Table 2.
Effect of initial concentration of AgNO3 on the loading
Table 2.
Effect of initial concentration of AgNO3 on the loading
Initial concentration of AgNO3 (mg/L) Crosslinker (%) Loading (mg/L) Loading (%) 110 30 38.6 35.1 438 30 134 30.6 767 30 193 25.1 1096 30 232 21.2 110 45 24 21.8 438 45 59 13.5 767 45 75 9.8 1096 45 93 8.5 Table 2 also illustrates that the silver ion loading in the membrane matrix decreases with increasing the crosslink density. The reason is that with increasing the crosslink density, the swelling and diffusion of silver ion from solution into the matrix network is reduced.
Thermal Analysis
TGA and DTG methods were used to thermally characterize the crosslinked-chitosan in comparison to the intact polysaccharide (Fig. 19). The initial weight loss up to about 120 ℃ is due to the presence of moisture. A rapid weight loss of about 30%-35% is observed for pure chitosan between 260 and 340 ℃ and this may be attributed to the splitting of the polysaccharide chains. These results were in accordance with previous reports[53]. For the crosslinked-chitosan and the silver nanocomposite the weight loss was observed at a former stage, i.e. between 230 and 330 ℃. Figure 19 also shows that about 88% weight loss takes place at 600 ℃ for chitosan, and it can degrade completely at 700 ℃. However, for crosslinked-chitosan and silver nanocomposite, a residual weight of nearly 25% was observed at this temperature. The high char yield, up to 18 wt%, at 700 ℃ observed in thermogram indicates that crosslinked-chitosan and silver nanocomposite have higher thermal stability than that of chitosan (char yield=1.2%). This stability caused by the strong bonding between the polymer chains in crosslinked-chitosan.
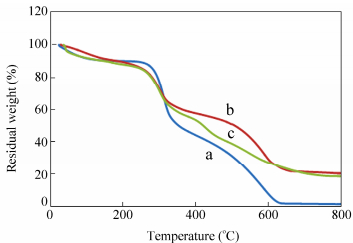 图19
Thermogravimetric analysis (TGA) of (a) chitosan, (b) crosslinked-chitosan and (c) crosslinked-chitosan silver nanocomposite
Figure19.
Thermogravimetric analysis (TGA) of (a) chitosan, (b) crosslinked-chitosan and (c) crosslinked-chitosan silver nanocomposite
图19
Thermogravimetric analysis (TGA) of (a) chitosan, (b) crosslinked-chitosan and (c) crosslinked-chitosan silver nanocomposite
Figure19.
Thermogravimetric analysis (TGA) of (a) chitosan, (b) crosslinked-chitosan and (c) crosslinked-chitosan silver nanocomposite
Figure 20 demonstrates that the rate of thermal degradation of pure chitosan at 300 ℃ is higher than that of crosslinked-chitosan and the silver nanocomposite. Reducing the thermal stability of silver nanocomposite between 350 and 600 ℃ compared with crosslinked-chitosan, can be due to the high thermal conductivity and heat transfer of the nanosilver.
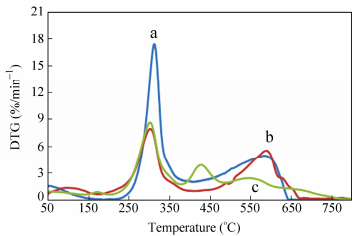 图20
DTG analysis of (a) chitosan, (b) crosslinked-chitosan and (c) crosslinked-chitosan silver nanocomposite
Figure20.
DTG analysis of (a) chitosan, (b) crosslinked-chitosan and (c) crosslinked-chitosan silver nanocomposite
图20
DTG analysis of (a) chitosan, (b) crosslinked-chitosan and (c) crosslinked-chitosan silver nanocomposite
Figure20.
DTG analysis of (a) chitosan, (b) crosslinked-chitosan and (c) crosslinked-chitosan silver nanocomposite
Swelling in Salt Solutions
The osmotic pressure difference between the polymer network and aqueous phases decreases in salt solutions, resulting in decreasing the amounts of the swelling[45]. With multivalent cations, “ionic crosslinking”leads to an appreciably decrease in the swelling capacity. As indicated in Fig. 13, the swelling capacity slightly decreases for CaCl2 probably due to ionic crosslinking, but it is considerably increased for AlCl3. Aluminum chloride dissociates in water that forms an aluminum hexa-aqua complex ion, Al(H2O)63+. Partial hydrolysis of the aluminum ion in water produces an acidic solution (pH ≈ 3.8). As a result, the repulsion between the―NH2+ groups on the chitosan-chitosan chains can increase the swelling. Similar behavior was reported by our research group, previously[46].
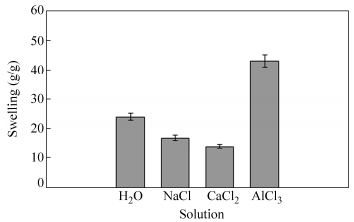 图13
Swelling capacity variation of the crosslinked-chitosan membrane (with 30% crosslinker) in saline solutions (0.15 mol/L)
Figure13.
Swelling capacity variation of the crosslinked-chitosan membrane (with 30% crosslinker) in saline solutions (0.15 mol/L)
图13
Swelling capacity variation of the crosslinked-chitosan membrane (with 30% crosslinker) in saline solutions (0.15 mol/L)
Figure13.
Swelling capacity variation of the crosslinked-chitosan membrane (with 30% crosslinker) in saline solutions (0.15 mol/L)
Swelling and Deswelling Kinetics
The swelling kinetics of the membranes was measured in consecutive time intervals at pH=2. Figure 17 represents the dynamic swelling behavior of the optimally prepared membranes. At first, the rate of the swelling rises and then it begins to flatten out. The equilibrium swelling was obtained after about 20 min. A power law style is concluded from the Fig. 17. The data may be well matched with a‘‘Voigt-based model’’Eq. (2)[52]:
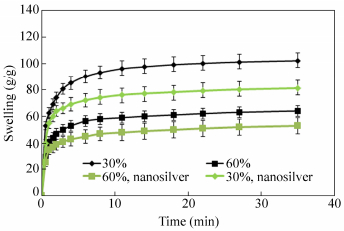 图17
Effect of the crosslinker weight ratio on the swelling kinetics of the membranes and the silver nanocomposites at pH=2
Figure17.
Effect of the crosslinker weight ratio on the swelling kinetics of the membranes and the silver nanocomposites at pH=2
图17
Effect of the crosslinker weight ratio on the swelling kinetics of the membranes and the silver nanocomposites at pH=2
Figure17.
Effect of the crosslinker weight ratio on the swelling kinetics of the membranes and the silver nanocomposites at pH=2
where St (g/g) is the swelling at time t, Se is the equilibrium swelling (power parameter, g/g), t is time for the swelling St, and t stand for the“rate parameter”. The rate parameters are found to be 75 and 45 s for crosslinked-chitosan with crosslinker weight ratio of 30% and 60%, respectively. The swelling of the silver nanocomposite was lower than that of the crosslinked-chitosan membrane. This is probably due to the more crosslinking points which was created by the silver nanoparticles inside the gel.
The deswelling water ratios of the crosslinked-chitosan with crosslinker weight ratio of 30% were measured using Eq. (1). Figure 18 shows the ratio of the remaining water as a function of time. As shown in this figure the deswelling water ratio of the sample indicates a weight reduction of about 32% at room temperature and about 73% at 50 ℃ from its original weight after 30 h. As a result the membranes have a good potential to hold the water at room temperature.
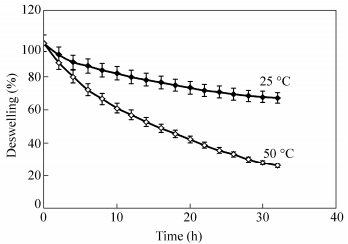 图18
Deswelling water ratio of the membranes with 30% weight ratio of crosslinker at 25 and 50 ℃
Figure18.
Deswelling water ratio of the membranes with 30% weight ratio of crosslinker at 25 and 50 ℃
图18
Deswelling water ratio of the membranes with 30% weight ratio of crosslinker at 25 and 50 ℃
Figure18.
Deswelling water ratio of the membranes with 30% weight ratio of crosslinker at 25 and 50 ℃
Degradation Test
Crosslinked-chitosan membranes showed a significant weight loss in acidic solution at room temperature (Fig. 10). The resulting polymer network contains acetal groups that can be hydrolyzed in acidic solution. After 12 h, the weight reduction of crosslinked-chitosan membranes at pH=4 and pH=2 was found to be 85.0% and 90.0%, respectively. However, the crosslinked-chitosan network is quite stable at neutral pH.
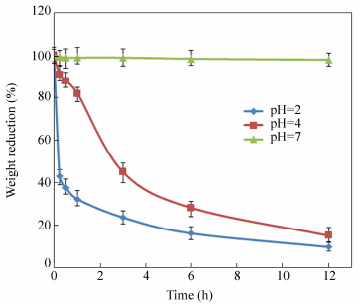 图10
Weight loss of crosslinked chitosan membranes with 60% weight ratio of crossliker at various pH
Figure10.
Weight loss of crosslinked chitosan membranes with 60% weight ratio of crossliker at various pH
图10
Weight loss of crosslinked chitosan membranes with 60% weight ratio of crossliker at various pH
Figure10.
Weight loss of crosslinked chitosan membranes with 60% weight ratio of crossliker at various pH
Synthesis and Characterization
Oxidation of the chitosan with periodate differs in many respects from the other polysaccharides because of the involvement of the amino groups. The behavior depends on the degree of N-acetylation. It has been reported that chitosan only partially oxidized by periodate and reaching degrees of oxidation around 0.5, when oxidizing with excess periodate[29]. In this work, chitosan was oxidized by sodium periodate to generate chitosan dialdehyde, which was used as a crosslinker. Degradation can also occur via cleavage of glycosidic bonds which results in depolymerization of the polysaccharide (Fig. 1). The aldehyde groups can react with primary amino groups of the chitosan to form imine bonds. Because the imine bonds are unstable and they can be readily hydrolyzed in aqueous solutions, a subsequent reduction by sodium borohydride stabilizes the conjugation (Fig. 2). The produced membranes before reduction and after reduction with sodium borohydride were depicted in Fig. 3.
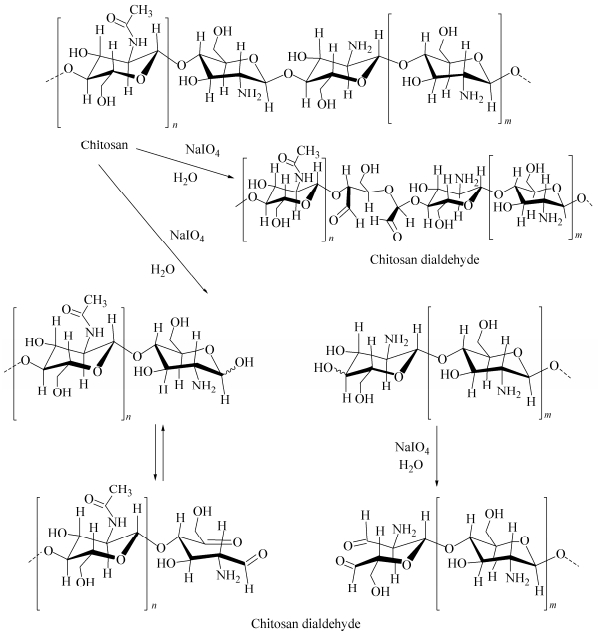 图1
Reaction of chitosan with sodium periodate and formation of the chitosan dialdehyde
Figure1.
Schematic representation of formation of crosslinked-chitosan
图1
Reaction of chitosan with sodium periodate and formation of the chitosan dialdehyde
Figure1.
Schematic representation of formation of crosslinked-chitosan
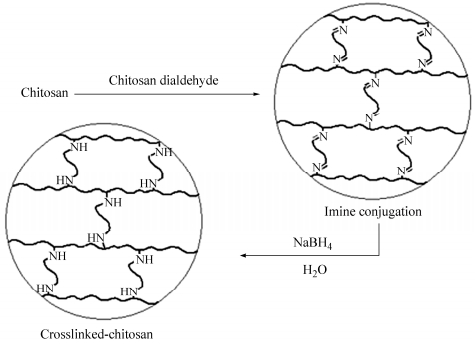 图2
Reaction of chitosan with sodium periodate and formation of the chitosan dialdehyde
Figure2.
Reaction of chitosan with sodium periodate and formation of the chitosan dialdehyde
图2
Reaction of chitosan with sodium periodate and formation of the chitosan dialdehyde
Figure2.
Reaction of chitosan with sodium periodate and formation of the chitosan dialdehyde
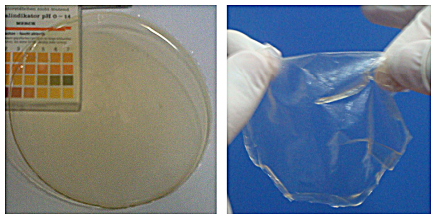 图3
Crosslinked-chitosan membranes before reduction (left) and after reduction with sodium borohydride (right)
Figure3.
Crosslinked-chitosan membranes before reduction (left) and after reduction with sodium borohydride (right)
图3
Crosslinked-chitosan membranes before reduction (left) and after reduction with sodium borohydride (right)
Figure3.
Crosslinked-chitosan membranes before reduction (left) and after reduction with sodium borohydride (right)
The FTIR spectra of the chitosan and partially oxidized chitosan are depicted in Fig. 4. The main characteristic absorption bands of the chitosan appear at 1656 cm-1 (C=O stretching), 1600 cm-1 (NH angular deformation), 3350-3450 cm-1 (OH hydroxyl group), and 1150-1040 cm-1 (C―O―C in glycosidic linkage), respectively (Fig. 4a). Figure 4(b) shows the IR spectrum of periodate oxidized chitosan after drying at room temperature. Generally, both the chitosan and the oxidized chitosan have similar peaks, but the oxidized chitosan has lower intensity of the absorption bands below 1500 cm-1. The decrease in peaks intensity can be attributed to the depolymerization of chitosan and the change in the number of hydrogen bonding. The intensity of the absorption bands also depends on the amount of the change in dipole moment associated with the vibration which can be higher for the chitosan because of smaller degree of freedom and mobility. Similar observation has been reported for high molecular weight dextran that was exposed to CO2 laser radiation[36].
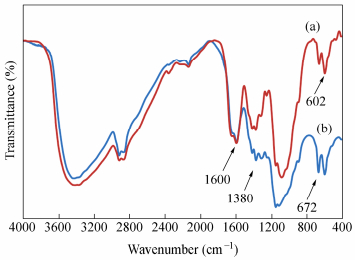 图4
FTIR spectra of (a) chitosan and (b) periodate oxidized chitosan
Figure4.
FTIR spectra of (a) chitosan and (b) periodate oxidized chitosan
图4
FTIR spectra of (a) chitosan and (b) periodate oxidized chitosan
Figure4.
FTIR spectra of (a) chitosan and (b) periodate oxidized chitosan
The FTIR spectra of the crosslinked-chitosan before and after reduction with sodium borohydride are indicated in Fig. 5. Figure 5(a) shows the appearance of a“shoulder”at 1646 cm-1, which can be attributed to imine C=N stretching. Figure 5(b) shows the FTIR spectra of the crosslinked-chitosan after reduction with sodium borohydride in aqueous solution. Absorbance at 1514 cm-1 and 1150 cm-1 can be attributed to secondary amine N―H bending and C―N stretching vibrations, respectively. It shows that the imine groups are converted to amine groups. Figure 5(c) demonstrates the FTIR spectrum of the silver nanocomposite. The interactions between crosslinked-chitosan chains and Ag-NPs are associated with the peak at 3425 cm-1. Broad peak is due to the presence of the interaction between the hydroxyl groups of chitosan and the partial positive charge on the surface the Ag-NPs. In addition, the stretching vibration at 1655 cm-1 corresponding to C=O group indicated that the silver nanoparticles are bounded to the functional groups present in chitosan.
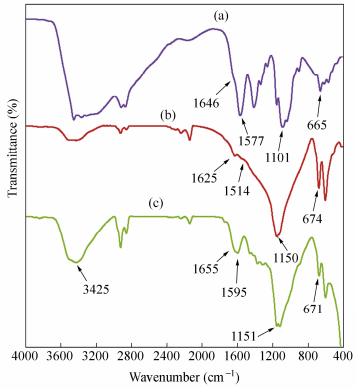 图5
FTIR spectra of (a) crosslinked-chitosan before reduction, (b) crosslinked-chitosan after reduction with sodium borhydride and (c) silver nanocomposite
Figure5.
FTIR spectra of (a) crosslinked-chitosan before reduction, (b) crosslinked-chitosan after reduction with sodium borhydride and (c) silver nanocomposite
图5
FTIR spectra of (a) crosslinked-chitosan before reduction, (b) crosslinked-chitosan after reduction with sodium borhydride and (c) silver nanocomposite
Figure5.
FTIR spectra of (a) crosslinked-chitosan before reduction, (b) crosslinked-chitosan after reduction with sodium borhydride and (c) silver nanocomposite
Figure 6 represents the 300 MHz 1H-NMR spectrum of chitosan and oxidized chitosan at room temperature. The assignment of chitosan peaks has already been reported in the literature[37, 38]. The NH2 proton was not observable in most cases as it is known to appear near the broad peak of water (δ=4.6-5.0). The data show that there is no considerable change in the ratio of integration values of the protons at δ=1.8 to that of the protons at δ=8.
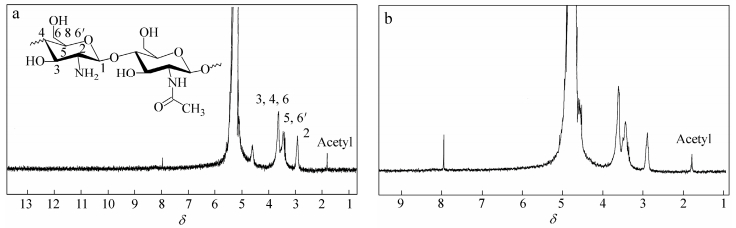 图6
The 1H-NMR spectra of (a) chitosan and (b) oxidized chitosan
Figure6.
The 1H-NMR spectra of (a) chitosan and (b) oxidized chitosan
图6
The 1H-NMR spectra of (a) chitosan and (b) oxidized chitosan
Figure6.
The 1H-NMR spectra of (a) chitosan and (b) oxidized chitosan
The elemental analysis data were used to confirm the product of oxidation in combination with FTIR and 1H-NMR spectral data. The data of elemental analysis are shown in Table 1. The results indicate that the amount of nitrogen decreases somewhat with increasing the concentration of peiodate solution at room temperature.
Table 1.
Elemental analysis data of chitosan and oxidized chitosan
Compound W(C) (%) W(H) (%) W(N) (%) CH 41.24 7.26 7.63 OC1 40.14 7.40 7.25 OC2 40.43 7.46 7.21 CH=chitosan, OC1=oxidized chitosan 1 (sodium periodate: 0.01 mol/L), OC2=oxidized chitosan 2 (sodium periodate: 0.03 mol/L) Figure 7 shows the dynamic viscosity of pure chitosan and oxidized chitosan. The viscosity and shear rate for the solutions were in the range of 0.001-0.1 Pa×s, and 10-1000 s-1, respectively. The corresponding shear stress is shown in Fig. 8.
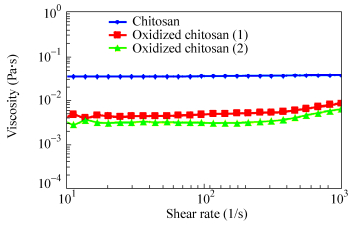 图7
Viscosity versus shear rate for chitosan, and oxidized chitosan (Oxidized chitosan 1: chitosan 0.015 g/mL, sodium periodate 0.01 mol/L, total volume of solution 20 mL, Oxidized chitosan 2: chitosan 0.015 g/mL, sodium periodate 0.03 mol/L, total volume of solution 20 mL)
Figure7.
Viscosity versus shear rate for chitosan, and oxidized chitosan (Oxidized chitosan 1: chitosan 0.015 g/mL, sodium periodate 0.01 mol/L, total volume of solution 20 mL, Oxidized chitosan 2: chitosan 0.015 g/mL, sodium periodate 0.03 mol/L, total volume of solution 20 mL)
图7
Viscosity versus shear rate for chitosan, and oxidized chitosan (Oxidized chitosan 1: chitosan 0.015 g/mL, sodium periodate 0.01 mol/L, total volume of solution 20 mL, Oxidized chitosan 2: chitosan 0.015 g/mL, sodium periodate 0.03 mol/L, total volume of solution 20 mL)
Figure7.
Viscosity versus shear rate for chitosan, and oxidized chitosan (Oxidized chitosan 1: chitosan 0.015 g/mL, sodium periodate 0.01 mol/L, total volume of solution 20 mL, Oxidized chitosan 2: chitosan 0.015 g/mL, sodium periodate 0.03 mol/L, total volume of solution 20 mL)
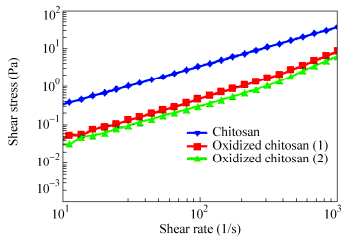 图8
Shear stress versus shear rate for chitosan, and oxidized chitosan (Oxidized chitosan 1: chitosan 0.015 g/mL, sodium periodate 0.01 mol/L, total volume of solution 20 mL; Oxidized chitosan 2: chitosan 0.015 g/mL, sodium periodate 0.03 mol/L, total volume of solution: 20 mL)
Figure8.
Shear stress versus shear rate for chitosan, and oxidized chitosan (Oxidized chitosan 1: chitosan 0.015 g/mL, sodium periodate 0.01 mol/L, total volume of solution 20 mL; Oxidized chitosan 2: chitosan 0.015 g/mL, sodium periodate 0.03 mol/L, total volume of solution: 20 mL)
图8
Shear stress versus shear rate for chitosan, and oxidized chitosan (Oxidized chitosan 1: chitosan 0.015 g/mL, sodium periodate 0.01 mol/L, total volume of solution 20 mL; Oxidized chitosan 2: chitosan 0.015 g/mL, sodium periodate 0.03 mol/L, total volume of solution: 20 mL)
Figure8.
Shear stress versus shear rate for chitosan, and oxidized chitosan (Oxidized chitosan 1: chitosan 0.015 g/mL, sodium periodate 0.01 mol/L, total volume of solution 20 mL; Oxidized chitosan 2: chitosan 0.015 g/mL, sodium periodate 0.03 mol/L, total volume of solution: 20 mL)
The rheological measurements show that the viscosity of chitosan solution decreases immediately by adding sodium periodate, indicating that depolymerization of chitosan can take place rapidly in the presence of periodate. Furthermore, the viscosity of the solution decreases with increasing the concentration of periodate solution. Similar results have been reported by Vold et al., indicating that the viscosity of the product depends on the reaction time and the concentration of periodate[33].
pH-dependent Swelling of the Membranes
The swelling behavior of the membranes was examined at various pH values between 2.0 and 12.0 at room temperature (Fig. 9). To prepare the pH media, standard HCl and NaOH solutions were diluted with distilled water to reach the desired acidic and basic pHs, respectively. The swelling decreases with increasing pH from pH=2 to pH=12. Swelling behavior is controlled mainly by amino groups (―NH2) on the C-2 carbon of the chitosan. Chitosan is a weak base with an inherent pKa of about 6.5. So, it is protonated at acidic pH, which increases charge density on the polymer, resulting in enhancing the osmotic pressure inside the network. The osmotic pressure difference between the internal and external solutions of the network is balanced by the swelling of the network.
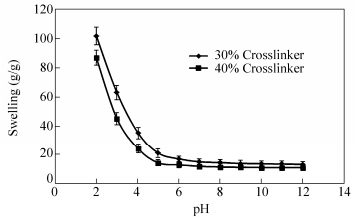 图9
pH-dependent swelling of the membranes
Figure9.
pH-dependent swelling of the membranes
图9
pH-dependent swelling of the membranes
Figure9.
pH-dependent swelling of the membranes
Effect of the Crosslinker Concentration
The effects of the crosslinker concentration on the swelling and gel content are shown in Fig. 11. The swelling capacity decreases with increasing the crosslinker concentration. Higher crosslinker concentration produces more crosslinked points in polymeric chains and increases the extent of the polymer network, which results in the development of the gel content. Similar behaviors have been reported in the literature[39, 40].
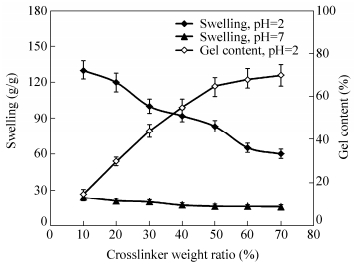 图11
Effect of the crosslinker concentration on water absorbency and sol content of the crosslinked-chitosan
Figure11.
Effect of the crosslinker concentration on water absorbency and sol content of the crosslinked-chitosan
图11
Effect of the crosslinker concentration on water absorbency and sol content of the crosslinked-chitosan
Figure11.
Effect of the crosslinker concentration on water absorbency and sol content of the crosslinked-chitosan
Figure 12 shows the stress-strain curves of the crosslinked chitosan films in dry state with different percentages of crosslinker. With increasing the crosslinker weight ratio the modulus values were increased, indicating that the stiffness of the membranes was influenced by the crosslinking level. Also, plastic deformation (deviation from linearity), elongation at break, and tensile strength increases with increasing the crosslinker weight ratio. Therefore, crosslinking improved the stress-strain relations and mechanical properties of the films. The possibility of using crosslinkers such as glutaraldehyde[41], genipin[42], and dialdehyde starch[43], to improve mechanical properties of chitosan has been explored. It has also been demonstrated that compared with the pristine chitosan film, the tensile strength of graphene oxide-chitosan film increases by 2.5 folds, from 32.7 MPa to 82.0 MPa[44].
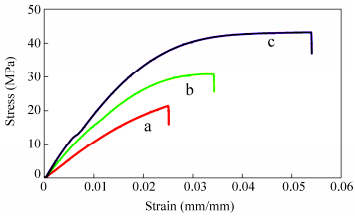 图12
Stress-strain curves of the crosslinked chitosan films with (a) 30%, (b) 60%, and (c) 90% weight ratio of chitosan dialehyde
Figure12.
Stress-strain curves of the crosslinked chitosan films with (a) 30%, (b) 60%, and (c) 90% weight ratio of chitosan dialehyde
图12
Stress-strain curves of the crosslinked chitosan films with (a) 30%, (b) 60%, and (c) 90% weight ratio of chitosan dialehyde
Figure12.
Stress-strain curves of the crosslinked chitosan films with (a) 30%, (b) 60%, and (c) 90% weight ratio of chitosan dialehyde
Antibacterial Properties of the Silver Nanocomposites
Among the metal nanoparticles, nanosilver displays reasonable antimicrobial activity[54-56]. The antibacterial properties of the silver nanocomposits were examined on E. coli, Pseudomonas aeruginosa, and Staphylococcus aureus (Fig. 21). The nitrofurantoin antibiotic was used as control. The silver nanocomposites exhibit an inhibition zone whereas the crosslinked-chitosan film does not have any effect on E. coli, P. aeruginosa and S. aureus. The antibacterial activity is resulting mainly from the release of silver nanoparticles from the silver nanocomposites. This could be attributed to the fact that silver nanoparticles can interact with sulfur and phosphorus containing compounds in the cells. The growth of inhibition ring was about 8.5 and 20 mm for the silver nanocomposite and nitrofurantoin, respectively. No inhibition zone was observed for nitrofurantoin antibiotic against P. aeruginosa.
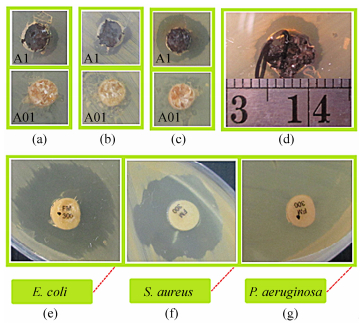 图21
Antibacterial effect on E. coli (a), S. aureus (b), and P. aeruginosa (c), for crosslinked-chitosan films (A1) and crosslinked-chitosan silver nanocomposite (A01); Comparison of the inhibition zone and antibacterial effect of the crosslinked-chitosan silver nanocomposite against P. aeruginosa (d), with the nitrofurantoin antibiotic against E. coli (e), S. aureus (f), and P. aeruginosa (g)
Figure21.
Antibacterial effect on E. coli (a), S. aureus (b), and P. aeruginosa (c), for crosslinked-chitosan films (A1) and crosslinked-chitosan silver nanocomposite (A01); Comparison of the inhibition zone and antibacterial effect of the crosslinked-chitosan silver nanocomposite against P. aeruginosa (d), with the nitrofurantoin antibiotic against E. coli (e), S. aureus (f), and P. aeruginosa (g)
图21
Antibacterial effect on E. coli (a), S. aureus (b), and P. aeruginosa (c), for crosslinked-chitosan films (A1) and crosslinked-chitosan silver nanocomposite (A01); Comparison of the inhibition zone and antibacterial effect of the crosslinked-chitosan silver nanocomposite against P. aeruginosa (d), with the nitrofurantoin antibiotic against E. coli (e), S. aureus (f), and P. aeruginosa (g)
Figure21.
Antibacterial effect on E. coli (a), S. aureus (b), and P. aeruginosa (c), for crosslinked-chitosan films (A1) and crosslinked-chitosan silver nanocomposite (A01); Comparison of the inhibition zone and antibacterial effect of the crosslinked-chitosan silver nanocomposite against P. aeruginosa (d), with the nitrofurantoin antibiotic against E. coli (e), S. aureus (f), and P. aeruginosa (g)
Table 3 exhibits the minimal inhibitory concentration (MIC) and the minimal bactericidal concentration (MBC) of the crosslinked-chitosan silver nanocomposite. The MBC/MIC ratio is a parameter that reflects the bactericidal capacity of the analyzed compounds. The MBC/MIC ratio of 2.0, 2.0, and 1.0 was achieved for E. coli, S. aureus, and P. aeruginosa, respectively. The results demonstrates that S. aureus and E. coli are more resistant to silver nanoparticles compared to P. aeruginosa, indicating that the bactericidal properties of the silver nanocomposite is affected by the type of the microorganism. The results also showed that the silver nanocomposites are good antibacterial materials regardless of the kind of bacteria used.
Table 3.
MIC and MBC of the silver nanocomposite for E. coli, S. aureus, and P. aeruginosa
Culture ATCC MIC (μg/mL) MBC (μg/mL) MBC/MIC ratio E. coli 8739 15.6 31.25 2.0 S. auresus 6538 15.6 31.25 2.0 P. aeruginosa 9027 15.6 15.6 1.0 CONCLUSIONS
Full-polysaccharide crosslinkged-chitosan membranes were prepared by crosslinking of chitosan with chitosan dialdehyde and subsequent reductive amination. Experimental studies indicate that degradation and depolymerization of chitosan can take place in the presence of periodate solution. Crosslinking also improved the stress-strain relations and mechanical properties of the films. The membranes will most probably possess high biodegradability and low toxicity, because no synthetic and toxic materials are used in the synthetic process. The silver nanocomposite was prepared via in situ reduction of silver nitrate (AgNO3) using sodium borohydride (NaBH4) as a reducing agent. TEM images indicate that crosslinked-chitosan nanocomposite comprises well defined silver nanoparticles with diameters in the range of 4-10 nm. The silver ion loading increases with increasing the silver ion concentration, and decreasing the crosslink density. The silver nanocomposites demonstrate good antibacterial effects against both Gram-positive and Gram-negative bacteria. The MBC/MIC ratio of 2.0, 2.0, and 1.0 was achieved for E. coli, S. aureus, and P. aeruginosa, respectively. The silver nanocomposites have potential applications in biomedical applications, and antibacterial wound-dressing.
-
-
[1]
Bhattarai, N., Gunn, J. and Zhang, M., Adv. Drug Deliv. Rev., 2010, 62(1):83 doi: 10.1016/j.addr.2009.07.019
-
[2]
Koyamatsu, Y., Hirano, T., Kakizawa, Y., Okano, F., Takarada, T. and Maeda, M., J. Control. Release, 2014, 173(1), 89
-
[3]
Armentano, I., Dottori, M., Fortunati, E., Mattioli, S. and Kenny, J.M., Polym. Degrad. Stab., 2010, 95(11):2126 doi: 10.1016/j.polymdegradstab.2010.06.007
-
[4]
Li, C., Zhang, J., Zu, Y.J., Nie, S.F., Cao, J., Wang, Q., Nie, S. P., Deng, Z.Y., Xie, M.Y. and Wang, S., Chin. J. Nat. Med., 2015, 13(9):641
-
[5]
Emam, H.E. and Ahmed, H.B., Carbohydr. Polym., 2016, 135:300 doi: 10.1016/j.carbpol.2015.08.095
-
[6]
Jain, A., Gupta, Y. and Jain, S.K., J. Pharm. Pharmaceut. Sci., 2007, 10(1):86
-
[7]
Luo, Y. and Wang, Q., Int. J. Biol. Macromol., 2014, 64:353 doi: 10.1016/j.ijbiomac.2013.12.017
-
[8]
Matricardi, P., Di Meo, C., Coviello, T., Hennink, W.E. and Alhaique, F., Adv. Drug Deliv. Rev., 2013, 65(9):1172 doi: 10.1016/j.addr.2013.04.002
-
[9]
Senel, S. and Mc Clure, S.J., Adv. Drug Deliv. Rev., 2004, 56(10):1467 doi: 10.1016/j.addr.2004.02.007
-
[10]
Aranaz, I., Harris, R. and Heras, A., Curr. Org. Chem., 2010, 14:308
-
[11]
Alvarez-Lorenzo, C., Blanco-Fernandez, B., Puga, A.M. and Concheiro, A., Adv. Drug Deliv. Rev., 2013, 65(9):1148 doi: 10.1016/j.addr.2013.04.016
-
[12]
Ngo, D.H., Vo, T.S., Ngo, D.N., Kang, K.H., Je, J.Y., Pham, H.N.D., Byun, H.G. and Kim, S.K., Food Hydrocolloid., 2015, 51:200 doi: 10.1016/j.foodhyd.2015.05.023
-
[13]
Holme, H.K., Davidsen, L., Kristiansen, A. and Smidsrod, O., Carbohydr. Polym., 2008, 73(4):656 doi: 10.1016/j.carbpol.2008.01.007
-
[14]
Wan Ngah, W.S., Hanafiah, M.A. and Yong, S.S., Colloids Surf B:Biointerfaces, 2008, 65(1):18 doi: 10.1016/j.colsurfb.2008.02.007
-
[15]
Emami Meybodi, Z., Imani, M. and Atai, M., Carbohydr. Polym., 2013, 92(2):1792 doi: 10.1016/j.carbpol.2012.11.030
-
[16]
Singh, A., Narvi, S.S., Dutta, P.K. and Pandey, N.D., Bull. Mater. Sci., 2006, 29(3):233 doi: 10.1007/BF02706490
-
[17]
Du, W.L., Niu, S.S., Xu, Z.R. and Xu, Y.L., J. Appl. Polym. Sci., 2009, 111(6):2881 doi: 10.1002/app.v111:6
-
[18]
Kulkarni, V.H., Kulkarni, P.V. and Keshavayya, J., J. Appl. Polym. Sci., 2007, 103(1):211 doi: 10.1002/(ISSN)1097-4628
-
[19]
Monier, M. and El-Sokkarya, A.M., Int. J. Biol. Macromol., 2010, 47(2):207 doi: 10.1016/j.ijbiomac.2010.04.020
-
[20]
Vargasa, G., Acevedob, J.L., Lopeza, J. and Romero, J., Mater. Lett., 2008, 62:3656 doi: 10.1016/j.matlet.2008.04.020
-
[21]
Hwang, H.D., Cho, H.J., Balakrishnan, P., Chung, C.W., Yoon, I.S., Oh, Y.K., Byun, Y. and Kim, D.D., Colloids Surf B:Biointerfaces, 2012, 91:106 doi: 10.1016/j.colsurfb.2011.10.052
-
[22]
Pujana, M.A., Perez-Alvarez, L., Iturbe, L.C. and Katime, I., Carbohydr. Polym., 2014, 101(1):113
-
[23]
Mekhail, M., Jahan, K. and Tabrizian, M., Carbohydr. Polym., 2014, 108:91 doi: 10.1016/j.carbpol.2014.03.021
-
[24]
Li, Q., Wang, X., Lou, X., Yuan, H., Tu, H., Li, B. and Zhang, Y., Carbohydr. Polym., 2015, 130:166 doi: 10.1016/j.carbpol.2015.05.039
-
[25]
Pujana, M.A., Perez-Alvarez, L., Cesteros Iturbe, L.C. and Katime, I., Carbohydr. Polym., 2013, 94(2):836 doi: 10.1016/j.carbpol.2013.01.082
-
[26]
Baran, E.T., Mano, J.F. and Reis, R.L., J. Mater. Sci. Mater. Med., 2004, 15(7):759 doi: 10.1023/B:JMSM.0000032815.86972.5e
-
[27]
Kimura, S., Isobe, N., Wada, M., Kuga, S., Ko, J.H. and Kim, U.J., Carbohydr. Polym., 2011, 83(4):1850 doi: 10.1016/j.carbpol.2010.10.049
-
[28]
Paradossi, G., Chiessi, E., Cavalieri, F., Moscone, D. and Crescenzi, V., Polym. Gels. Networks, 1998, 5(6):525 doi: 10.1016/S0966-7822(97)00026-9
-
[29]
Liu, G., Shi, Z., Kuriger, T., Hanton, L.R., Simpson, J., Moratti, S.C., Robinson, B.H., Athanasiadis, T., Valentine, R., Wormald, P.G. and Robinson, S., Macromol. Symp., 2009, 279(1):151 doi: 10.1002/masy.v279:1
-
[30]
Tan, H., Wu, B., Li, Ch., Mu, Ch., Li, H. and Lin, W., Carbohydr. Polym., 2015, 129:17 doi: 10.1016/j.carbpol.2015.04.029
-
[31]
Ge, L., Li, X., Zhang, R., Yang, T., Ye, X., Li, D. and Mu, Ch., Food Hydrocolloids, 2015, 51:129 doi: 10.1016/j.foodhyd.2015.04.029
-
[32]
Zahran, M.K., Ahmed, H.B. and El-Rafie, M.H., Carbohydr. Polym., 2014, 108:145 doi: 10.1016/j.carbpol.2014.03.005
-
[33]
Kozicki, M., Kołodziejczyk, M., Szynkowska, M., Pawlaczyk, A., Lesniewska, E., Matusiak, A., Adamus, A. and Karolczak, A., Carbohydr. Polym., 2016, 140:74 doi: 10.1016/j.carbpol.2015.12.017
-
[34]
Pourjavadi, A., Ghasemzadeh, H. and Mojahedi, F., J. Appl. Polym. Sci., 2009, 113(6):3442 doi: 10.1002/app.v113:6
-
[35]
Vold, I.M. and Christensen, B.E., Carbohydr. Res., 2005, 340(4):679 doi: 10.1016/j.carres.2005.01.002
-
[36]
Dlugunovich, V.A., Zhbankov, R.G., Zhdanovskii, V.A., Zagorskaya, S.A. and Firsov, S.P., J. Appl. Spect., 2006, 73:178 doi: 10.1007/s10812-006-0055-7
-
[37]
Hirai, A., Odani, H. and Nakajima, A., Polym. Bull., 1991, 26(1):87 doi: 10.1007/BF00299352
-
[38]
Lavertu, M., Xia, Z., Serreqi, A.N., Berrada, M., Rodrigues, A., Wang, D., Buschmann, M.D. and Gupta, A.A., J. Pharma. Biomed. Anal., 2003, 32(6):1149 doi: 10.1016/S0731-7085(03)00155-9
-
[39]
Pourjavadi, A. and Ghasemzadeh, H., Polym. Eng. Sci., 2007, 47(9):1388 doi: 10.1002/(ISSN)1548-2634
-
[40]
Pourjavadi, A., Ghasemzadeh, H. and Soleyman, R., J. Appl. Polym. Sci., 2007, 105(5):2631 doi: 10.1002/(ISSN)1097-4628
-
[41]
Parida, U.K., Nayak, A.K., Binhani, B.K. and Nayak, P.L., J. Biomater. Nanobiotechnol., 2011, 2:414 doi: 10.4236/jbnb.2011.24051
-
[42]
Jin, J., Song, M. and Hourston, D.J., Biomacromolecules, 2004, 5(1):162 doi: 10.1021/bm034286m
-
[43]
Croisier, F. and Jerome, Ch., Eur. Polym. J., 2013, 49(4):780 doi: 10.1016/j.eurpolymj.2012.12.009
-
[44]
Zuo, P.P., Feng, H.F., Xu, Z.Z., Zhang, L.F., Zhang, Y.L., Xia, W. and Zhang, W.Q., Chem. Centr. J., 2013, 7(1):39 doi: 10.1186/1752-153X-7-39
-
[45]
Pourjavadi, A., Samadi, M. and Ghasemzadeh, H., Starch/Stärke, 2008, 60(2):79 doi: 10.1002/(ISSN)1521-379X
-
[46]
Pourjavadi, A., Aghajani, V. and Ghasemzadeh, H., J. Appl. Polym. Sci., 2008, 109(4):2648 doi: 10.1002/(ISSN)1097-4628
-
[47]
Oluwafemi, O.S., Lucwaba, Y., Gura, A., Masabeya, M., Ncapayi, V., Olujimi, O.O. and Songca, S.P., Colloids Surf B:Biointerfaces, 2013, 102:718 doi: 10.1016/j.colsurfb.2012.09.001
-
[48]
Sharma, V.K., Yngard, R.A. and Lin, Y., Adv. Colloid Interface Sci., 2009, 145(1-2):83 doi: 10.1016/j.cis.2008.09.002
-
[49]
Vimala, K., Samba Sivudu, K., Murali Mohan, Y., Sreedhar, B. and Mohana Raju, K., Carbohydr. Polym., 2009, 75(3):463 doi: 10.1016/j.carbpol.2008.08.009
-
[50]
Murali Mohan, Y., Lee, K., Premkumar, T. and Geckeler, K.E., Polymer, 2007, 48:158 doi: 10.1016/j.polymer.2006.10.045
-
[51]
Murthy, P.S.K., Murali Mohan, Y., Varaprasad, K., Sreedhar, B. and Mohana Raju, K., Colloid Interface Sci., 2008, 318(2):217 doi: 10.1016/j.jcis.2007.10.014
-
[52]
Zhou, W.J., Yao, K.J. and Kurth, M.J., J. Appl. Polym. Sci., 1996, 62(6):911 doi: 10.1002/(ISSN)1097-4628
-
[53]
Monier, M., Wei, Y., Sarhan, A.A. and Ayad, D.M., Polymer, 2010, 51(5):1002 doi: 10.1016/j.polymer.2010.01.001
-
[54]
Guo, L., Yuan, W., Lu, Z. and Li, C.M., Coll. Surf. A:Physicochem. Eng. Asp., 2013, 439:69 doi: 10.1016/j.colsurfa.2012.12.029
-
[55]
Mohamed, R.R. and Sabaa, M.W., Int. J. Biol. Macromol., 2014, 69:95 doi: 10.1016/j.ijbiomac.2014.05.025
-
[56]
Ouay, B.L. and Stellacci, F., Nano Today, 2015, 10:339 doi: 10.1016/j.nantod.2015.04.002
-
[1]
-

 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 952
- HTML全文浏览量: 13
 下载:
下载:
