
 Login In
Login In
2020 Volume 2 Issue 1
美国西北大学黄嘉兴教授、湖南大学周一歌教授及成都电子科技大学康毅进教授提出 “流动电催化”(Fluidized Electrocatalysis)策略,显著提高电催化剂的抗疲劳性能以及电催化反应的稳定性,甚至可以让很不稳定的催化剂达到持久稳定的催化效果。

在电催化反应中,催化剂材料通常被粘附在电极,比如碳电极的表面,而后浸入电解液中进行长时间、连续的电化学反应(图1a)。电催化反应有一些普遍的疲劳机制,例如反应中间体可能导致催化剂表面中毒,催化剂颗粒在长时间的电化学“压力”下发生团聚、烧结、溶解或钝化等。另外,反应活性物种到催化剂表面的扩散受限也会导致催化电流衰减。催化剂疲劳会大大降低催化剂的工作效率,缩短其寿命,导致整个电化学体系性能下降。针对此问题,传统的策略是从催化剂本身出发调控其表面状态和化学成分,或通过改进催化剂载体材料来防止催化剂颗粒的聚集和脱落,然而由于催化剂疲劳机制多种多样、催化剂及其载体的材料、成分和结构也各不相同,这些针对于催化材料本身的解决方案往往只适用于特定的催化剂,不具有普适性。
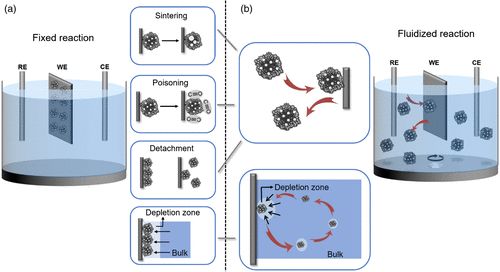
图1
在电催化过程中,电极反应只有电子转移步骤需要依赖电极。然而在常规催化体系中,由于电极一直处于极化状态,这会对催化剂产生不必要的额外的电化学压力,从而导致其疲劳和性能衰减。基于以上思考并结合单颗粒电分析化学的进展,美国西北大学黄嘉兴教授、湖南大学周一歌教授及成都电子科技大学康毅进教授合作,提出“流动电催化”的新策略来提高电催化剂的抗疲劳性能(图1b):催化剂颗粒并非以传统方式固定在电极上,而是在电解液中流动。
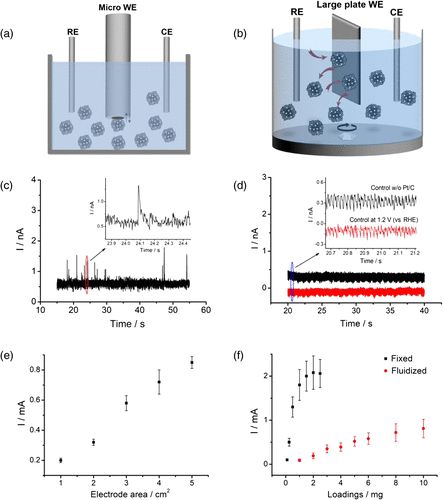
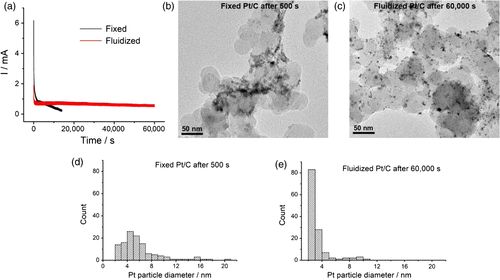

综上所述,该工作提出了流动电催化策略,将催化剂颗粒的工作模式由传统的长时间、连续性工作转变为轮流、间断性工作,避免了电化学压力的不断积累,同时,催化剂颗粒将经历更快的反应动力学并输出更高的电流效率,有利于抑制材料性能的衰减,提高催化剂长时间工作的稳定性。
当然,该流动策略的操作方式不可避免会带来体积能量密度的局限,但仍然可付诸大型固定电源供给与大规模电合成等实际应用场景。同时,流动催化剂较固定催化剂具有更高的稳定性,且易于回收及再利用,因此长时间的工作成本将远远低于固定催化剂。衡量体积能量密度与成本,该策略与改善、发展新型催化剂的实践可结合并行,有望发展成一种普适的提高电催化体系总体性能及稳定性的简单、高效的新方法。
该工作以封面文章形式发表在CCS Chemistry 2020年第一期,并于近期被美国化学会新闻周刊Chemical & Engineering News (C&EN)报道(https://cen.acs.org/synthesis/catalysis/Free-floating-electrocatalysts-outperform-tethered/98/web/2020/02)。牛津大学Richard G.Compton教授评价该工作为:“The work is groundbreaking in that it takes particle-impact experiments from the academic study of single nanoparticle electrocatalysis and suggests that they can be scaled up with considerable benefit.”
文章详情:
Fluidized Electrocatalysis
Yi-Ge Zhou, Yijin Kang, and Jiaxing Huang
Link: https://doi.org/10.31635/ccschem.020.201900065
Citation: CCS Chem. 2020, 2, 31–41
武汉大学付磊课题组设计了一种基于液态金属的保护层,首次利用液态金属的脆化和快速扩散等特性,显著抑制锂枝晶的生长,从而保护锂金属电池在高倍率下稳定长循环。
由于锂离子电池的能量密度已经难以满足日益增长的社会需求,具备更高能量密度的新一代电池在近年来受到越来越多的关注。其中,锂金属电池凭借锂金属电极超高理论比容量和极低电化学电位跻身前列。但是在长时间充放电过程中,锂金属电极的本征特性(包括极高反应活性和严重的体积膨胀)使其难以避免锂枝晶生长,而尖锐的枝晶极易刺穿隔膜,造成电池爆炸,这一严重的安全隐患也阻碍了锂金属电池的实际应用。
液态金属在新型电池中已经崭露头角,诺奖得主Goodenough教授在2016年提出钠钾合金作为室温液态金属电极。其流动性和自愈性能够抑制电极表面产生枝晶,但是这类材料的高反应活性、电极封装等问题仍亟待解决。那么针对现有蓬勃发展的锂金属电极,液态金属是否能为抑制枝晶生长给出另一份答卷呢?
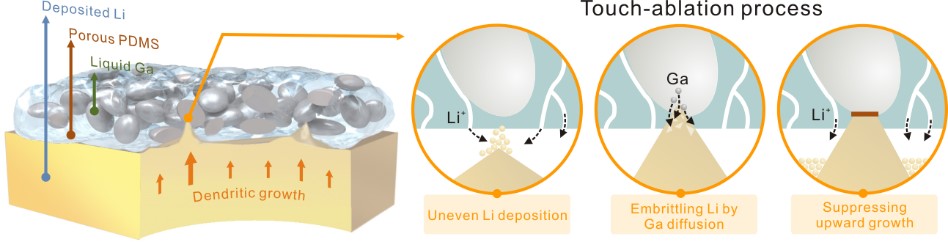
近期,武汉大学付磊课题组另辟蹊径,聚焦于典型的液态金属——镓(Ga),制备出Ga基复合膜作为锂金属负极的保护层。该组提出了接触−消融机制,当枝晶刺入保护层时,即可触发Ga发生独特的脆化现象,合金化接触区域,抑制枝晶向上生长的应力,从而实现均匀的锂沉积。通过精心设计和优化膜结构,将Ga液滴限域于分级多孔的聚二甲基硅氧烷(PDMS)膜中,仅2 μm的膜厚实现Ga的稳定过冷态(熔点降低至–16.6oC),使其在长循环中保持液态性质。同时,PDMS的电绝缘性避免Ga参与电化学反应,而多孔通道确保了锂离子的快速传输,提高电解液浸润性(图1)。
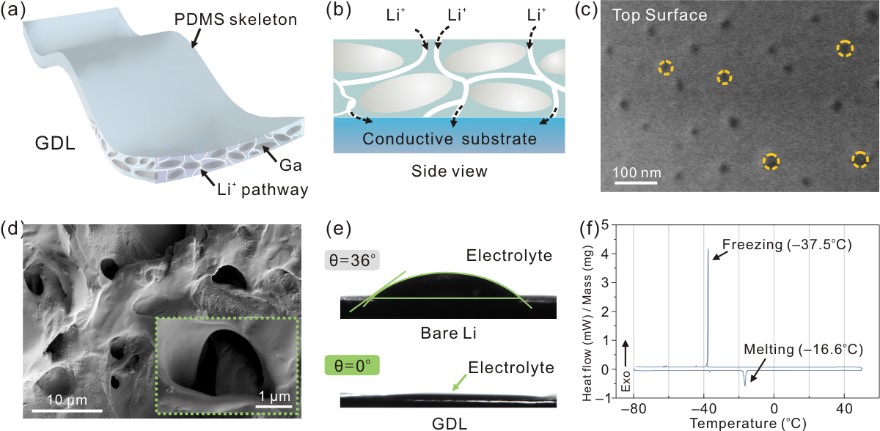
图1
通过X射线光电子能谱的深度分析,作者证实了合金化产物是Ga14Li3。这与Ga发生电化学反应的产物截然不同,且锂在该合金中处于特殊的非键合状态,有利于在循环过程中锂离子的可逆脱出。二次离子质谱验证了锂沉积集中于保护层下方,说明锂离子能通过保护层并在其下方实现均匀沉积(图2)。
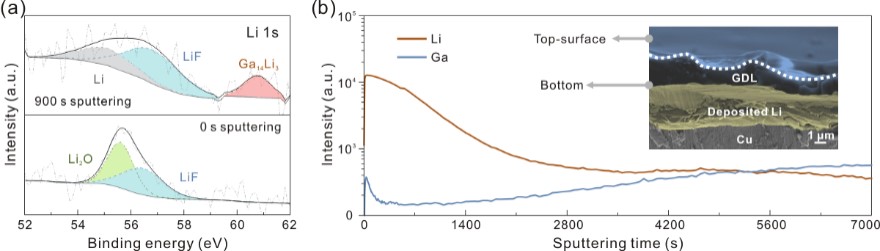
图2
作者进一步对比不同电流密度下对称电池的长循环性能来确认保护层的作用(图3)。在2 mA cm−2的电流密度下,未修饰的锂金属负极和纯PDMS修饰的锂负极均在较短时间内发生短路。而保护层作用下的锂电极寿命可超过2000 h。值得一提的是,即使在超高电流密度(10 mA cm−2)和大容量(5 mA h cm−2)下,受Ga基保护层作用的对称电池仍能维持极其稳定的电压曲线,这证实了接触−消融机制在高锂离子通量下的稳定性。此外,在保护层作用下,全电池也展现出优异的倍率性能和循环稳定性,说明了其实际应用的潜力。
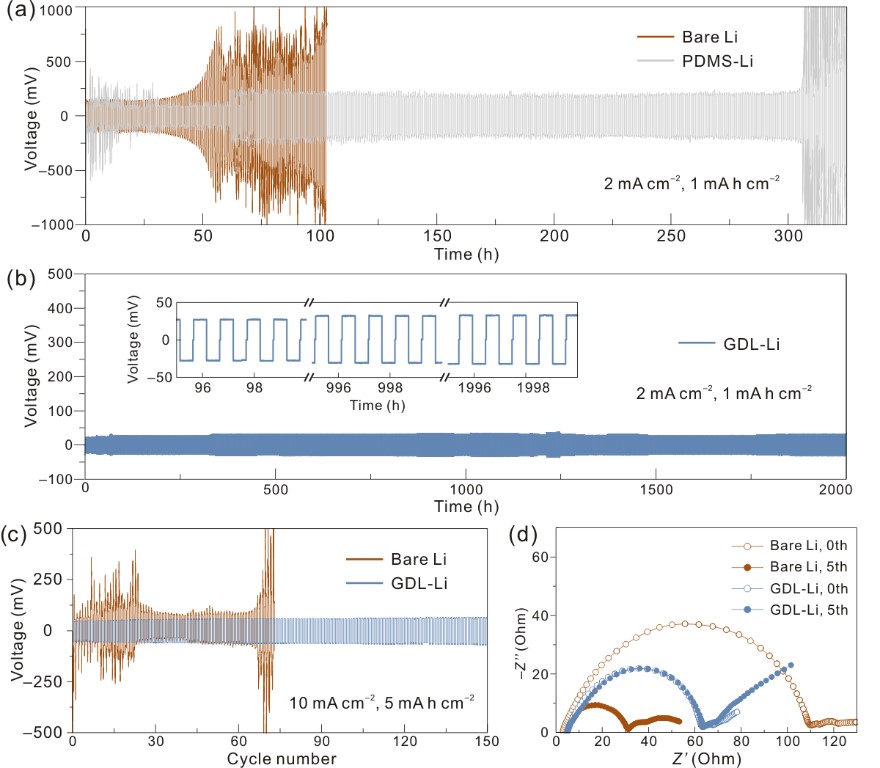
图3
综上所述,该研究工作提供了一种可以接触消融锂枝晶的保护层,为锂金属负极提供实时保护。这些结果强调了与枝晶直接反应的重要性,并且该保护层可与其他抑制手段强强联手,有望完全实现金属负极的无枝晶生长。该工作以research article 的形式发表在CCS Chemistry,并在CCS Chemistry官网“Just Published”栏目上线。
文章详情:
Touch Ablation of Lithium Dendrites via Liquid Metal for High-Rate and Long-Lived Batteries
Wenjie Wang†, Xiaohui Zhu†, and Lei Fu*
Citation: CCS Chem. 2020, 2, 686–695
Link:https://doi.org/10.31635/ccschem.020.202000182
中山大学夏炜、香港大学孙红哲课题组通过解析金黄色葡萄球菌醛基脱氢酶纯蛋白以及复合物晶体结构,发现其特有的“C-helix”结构域在结合特异性底物过程中发生的构象变化,提出了金黄色葡萄球菌醛基脱氢酶识别特异性底物的分子门控机制,并通过一系列生化实验辅助验证。
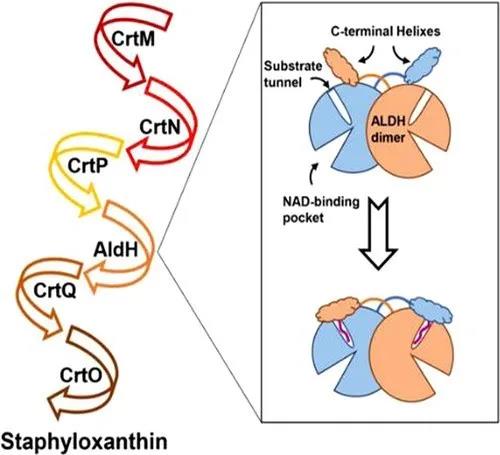
金黄色葡萄球菌是一种常见人类病原菌,可引起一系列感染性疾病,由于抗生素的滥用,产生了许多耐药性的金黄色葡萄球菌,而相对于开发新的抗生素,发展新的抗感染疗法对于对抗病毒感染更为有效。超过90%的金黄色葡萄球菌临床分离株会产生一种金黄色的含有30个碳(C30)的长链类胡萝卜素分子,称为葡萄球菌黄素,该色素可作为抗氧化剂提高细菌对于活性氧的耐受能力。因此,阻断其生物合成途径是一项重要的工作。
葡萄球菌黄素的生物合成路径需要一系列催化酶的参与(图1)。其中,醛基脱氢酶(SaAldH)是最近被发现的参与色素合成的酶类,其功能是催化长链不饱和醛4,4’- diaponeurosporen-4-al生成对应的羧酸。近期,中山大学夏炜课题组以及香港大学孙红哲课题组报道了SaAldH纯蛋白以及其与特异性底物复合物的晶体结构,在分子层面揭示了SaAldH识别特异性底物——多不饱和长碳链脂肪醛的门控机制。
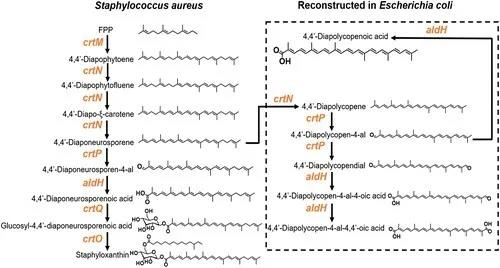
图1 葡萄球菌黄素合成通路示意图
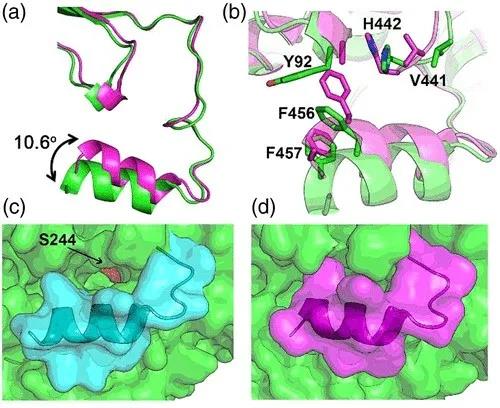
通过对SaAldH和特异性底物之间的相互作用进行分析,作者们发现特异性底物与SaAldH底物结合通道中的氨基酸残基形成广泛的疏水相互作用。另外,酶序列上Y116与底物的2-甲基形成π–σ相互作用,F457与底物末端的两个甲基形成疏水相互作用,暗示着“C-helix”同时也参与了对特异性底物的识别(图3)。
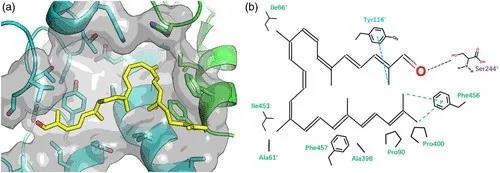
图3 SaAldH与其特异性底物相互作用分析
最后,作者们通过体外酶活实验、过氧化氢耐受实验以及巨噬细胞吞噬实验对关键氨基酸位点进行验证,证实结构中看到的关键性位点确实参与了特异性底物的识别(图4)。
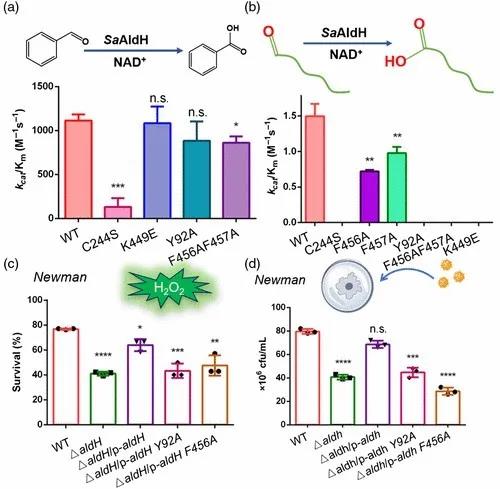
图4 关键氨基酸参与特异性底物识别的生化实验验证
此项研究得到了国家自然科学基金、香港研究资助局、中国教育部以及中央高校基础研究经费的资助。该工作以research article的形式发表在CCS Chemistry,并在官网“Just Published”栏目上线。
文章详情:
Structural Insight into the Substrate Gating Mechanism by Staphylococcus aureus Aldehyde Dehydrogenase
Xuan Tao , Zhemin Zhang , Xiao Zhang , Hongyan Li, Hongzhe Sun *, Zong-Wan Mao& Wei Xia *
Citation:CCS Chem. 2020, 2, 946–954
Link:https://doi.org/10.31635/ccschem.020.202000219
华东理工大学王俊有课题组报道了一种可控量化制备聚电解质纳米凝胶的通用方法,提出了静电组装导向聚合(Electrostatic Assembly Directed Polymerization)的技术途径,实现了一系列聚电解质纳米凝胶的高效、量化制备,为该类材料的实际应用奠定了基础。
聚电解质纳米凝胶是由聚电解质交联形成的水凝胶纳米粒子,其不仅具备常规纳米水凝胶的结构特征,同时还带有大量的电荷,可以高效地负载生物功能分子(酶、DNA,RNA、多肽等),并保持其(次级)结构和生物功能。因此,聚电解质纳米凝胶作为功能性“软”载体被广泛应用于药物传输、组织工程、生物传感、以及催化等不同领域。然而,聚电解质纳米凝胶的高效、量化制备仍然比较困难,发展新的合成策略是解决此问题的关键。
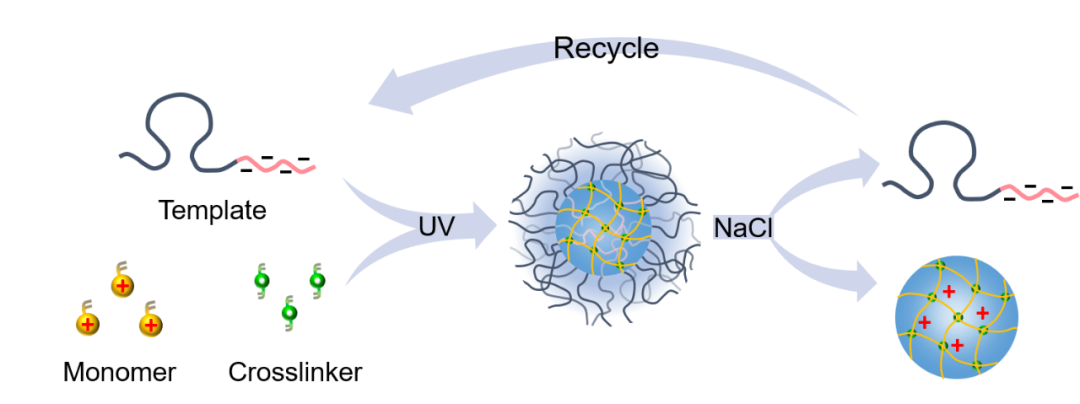
考虑到单体的电荷特性,该课题组在前期聚电解质可控组装的研究基础上(Angew. Chem. Int. Ed. 2018, 57, 12680;Angew. Chem. Int. Ed. 2019, 58, 8494),利用静电组装构筑限域环境,实施可控聚合,提出了静电组装导向聚合的方法,实现了不同聚电解质纳米凝胶的高效、量化制备。该方法利用聚离子-中性嵌段聚合物为模板,引发带相反电荷的单体与交联剂原位共聚,得到分散均一的复合胶束。反应完成后,利用高盐溶液脱除复合胶束中的嵌段聚合物,得到结构和尺寸高度可控的聚电解质纳米凝胶,且分离出的嵌段聚合物可再次作为模板重复循环使用(图1)。

图2 (a)不同正电单体合成的纳米凝胶的粒径分布;(b)不同氯化钠浓度下合成的纳米凝胶的粒径;(c)不同交联剂合成的PDMAEMA纳米凝胶的粒径分布;(d)不同单体浓度和体积下合成的PDMAEMA纳米凝胶的粒径分布。
静电组装导向聚合方法具有以下特点:1. 方法简便通用:水相自由基聚合,适用于多种正、负电荷单体,可以制备一系列聚电解质纳米凝胶(图2a)。2. 凝胶尺寸、结构可控:改变合成过程中的盐浓度可调控纳米凝胶尺寸(图2b),调控交联剂和交联度则可控制凝胶结构和性能(图2c)。3. 高浓度和体积下聚合可控,可量化制备聚电解质纳米凝胶(图2d)。4. 模板可回收循环使用。
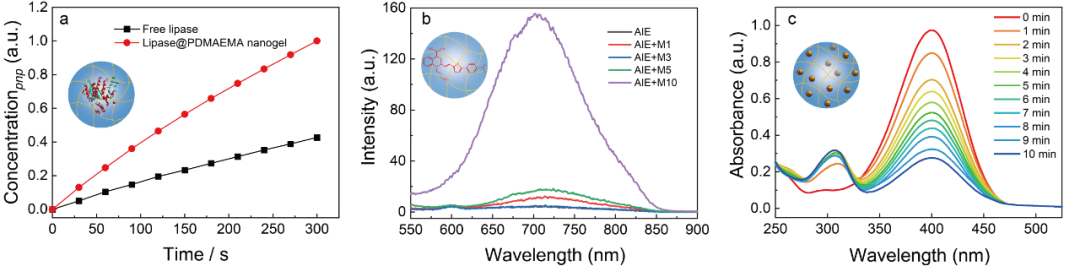
图3 (a)游离脂肪酶与负载于PDMAEMA纳米凝胶中脂肪酶的催化活性;(b)AIE分子在不同纳米凝胶中的荧光强度;(c)Au@PDMAEMA纳米凝胶催化对硝基苯酚过程中,紫外吸光度随时间的变化。
为验证聚电解质纳米凝胶作为“软”纳米载体的普适性应用,该团队将一系列功能性客体如脂肪酶、AIE荧光分子以及Au纳米颗粒负载于聚电解质纳米凝胶中,并研究了纳米凝胶的微环境对功能性客体性质的影响。研究发现,所制备的纳米凝胶能够有效地负载脂肪酶,并显著提升其催化活性(图3a);由于不同聚电解质纳米凝胶内部微环境存在差异,使得所负载的AIE分子表现出不同的荧光特性(图3b);此外,所制备的纳米凝胶对Au纳米颗粒亦展示出很好的负载能力,并可催化模型反应高效进行(图3c)。
综上所述,该研究工作展示了一种简单、高效、量化制备聚电解质纳米凝胶的新策略和新思路,将为聚电解质基“软”载体材料的实际应用提供重要的技术支撑。该工作以research article 的形式发表在CCS Chemistry上,论文的第一作者为华东理工大学博士研究生丁鹏,通讯作者为王俊有副教授。Cohen Stuart教授和郭旭虹教授在材料表征及数据分析上提供了帮助与支持。
文章详情:
Efficient and Generic Preparation of Diverse Polyelectrolyte Nanogels by Electrostatic Assembly Directed Polymerization
Peng Ding, Jianan Huang, Cheng Wei, Wei Liu, Wenjuan Zhou, Jiahua Wang, Mingwei Wang, Xuhong Guo, Martien A. Cohen Stuart & Junyou Wang*
Citation:CCS Chem. 2020, 2, 1016–1025
Link:https://doi.org/10.31635/ccschem.020.202000354
南方科技大学何凤课题组从分子设计入手,以嵌段共聚物为构筑基元,精确制备了基于共轭聚合物的菱形二维自组装结构。这些结构实现了半导体层与绝缘层厚度的精确分离,在外接电压下达到了可调控的隧穿效应。利用这种策略,成功制备了基于个体二维自组装体的压力传感器件,在微纳尺度下呈现了优异的灵敏度
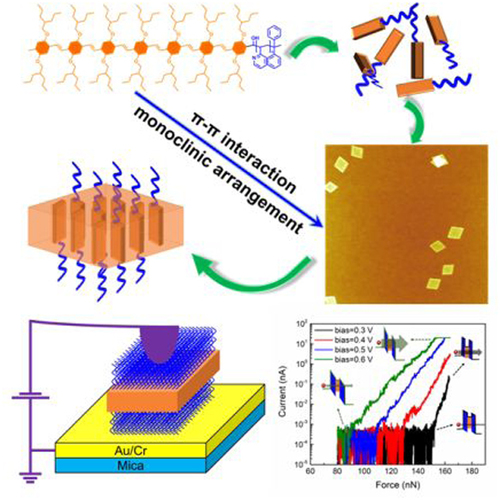
有机二维材料因其特殊的超薄扁平结构而具有独特的机械强度、电导性、透明度和柔韧性,被认为是生物传感器、电子和光电等微纳器件应用的最具潜力的候选材料。由于无序性的影响,二维有机材料大多表现的特性是其各组分性质的混合,这极大地限制了其应用。在微纳尺度下,为不同的应用目的制造具有不同特性的组分且有序排列的复杂结构是至关重要的科学课题。
近年来,通过嵌段共聚物(block copolymer,BCP)“自下而上”自组装来制备二维材料制备方法被建立和发展起来,而且成本低、效率高、质量高且可控。这种构筑基元由具有不同的成分组成,为实现微纳结构中可调的机械、光学和电学性质提供潜在的机会。然而,这些共聚物分子是如何自发地组装成二维形貌,以及它们在二维结构是如何排列堆积的,仍然是一个悬而未决的课题。因此,从分子设计的角度出发,制备精细可控的聚合物二维结构,并通过晶体学的修饰进一步调整其形貌是具有重要意义的。另外,尽管二维材料具有可以预见的优秀性能,但是目前对于聚合物二维材料的研究仍旧大多关注于形貌的制备与调控,真正有关其物理性能的研究还寥寥无几,这不得不说是一个遗憾。
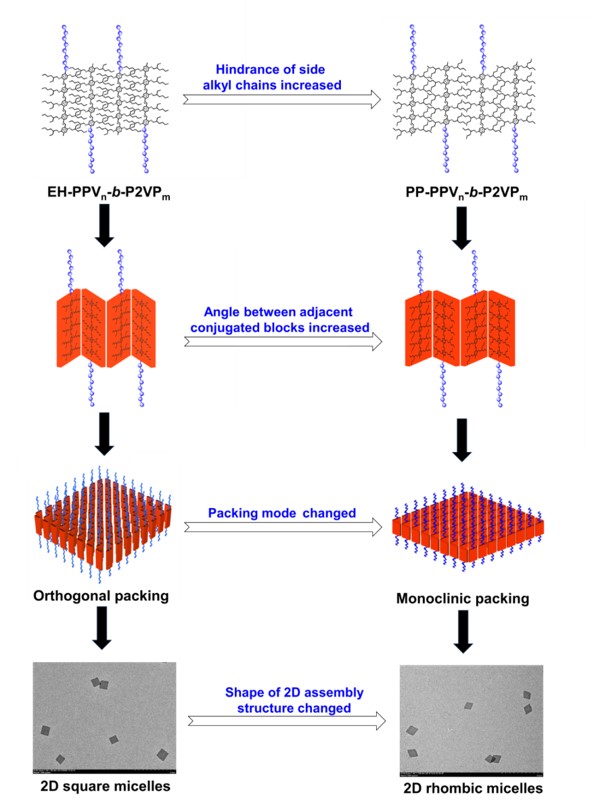
图1 通过改变共轭聚合物的侧边烷基链,利用晶体学调节的方法控制二维自组装结构的形状。
近期,南方科技大学的何凤课题组聚焦于具有半导体性质的共轭聚合物——聚对苯撑乙烯poly(p-phenylenevinylene) (PPV),以其作为成核嵌段成功了构筑嵌段共聚物。通过溶剂亲疏作用与分子间的π-π相互作用,在特定的溶剂中,这些嵌段共聚物可以形成规则均一的菱形二维自组装形貌。通过改变溶液浓度与分子嵌段比,可以实现形貌尺寸的精确控制。与该课题组之前的研究对比,可以发现通过调整共轭骨架两侧的烷基链,可以改变分子间π-π相互作用的空间位阻,不同的位阻导致分子堆积由正交排列转变为单斜排列,从而从宏观上观察到相似结构的嵌段共聚物形成的二维自组装形貌由正方形转变为菱形(图1)。这种二维自组装形貌的构筑策略是具有普适性的。
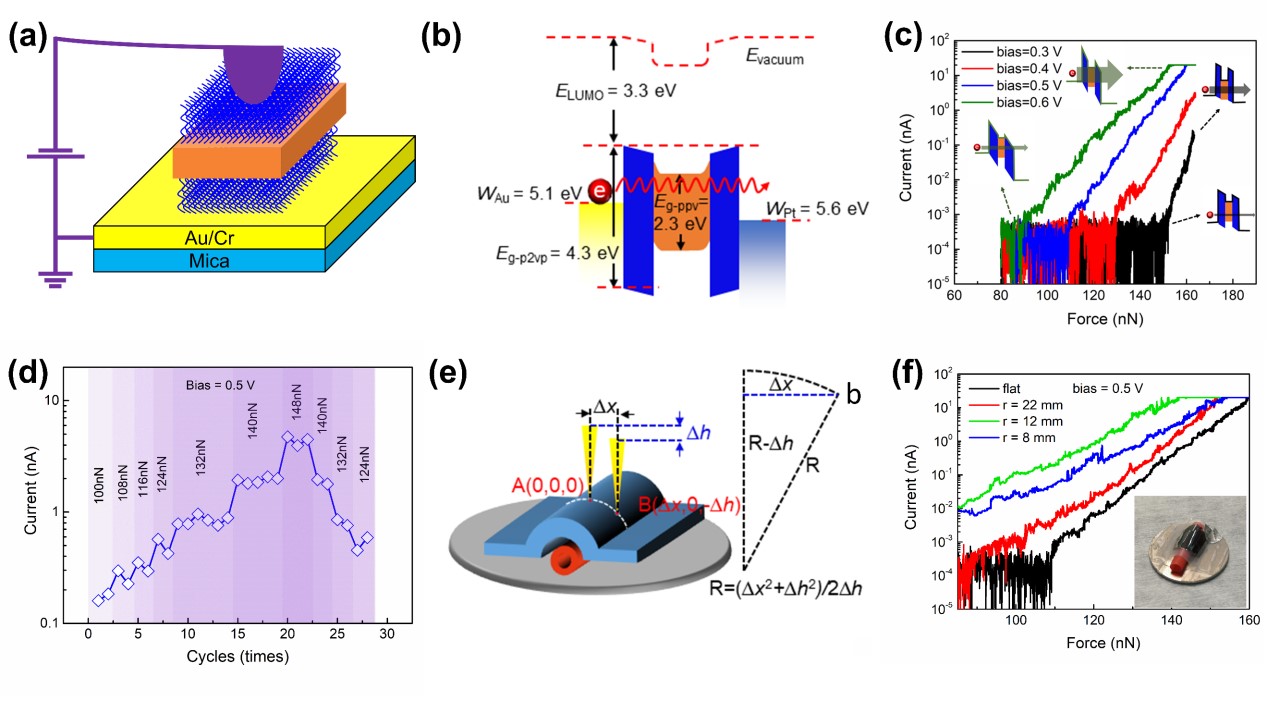
图2 基于二维自组装胶束构筑的隧穿效应压力传感器件性能研究。(a)隧穿效应压力传感器件结构;(b)隧穿结器件的能级结构;(c)不同压力下器件的电流-压力曲线;(d)隧穿器件的耐受性测试;(e)柔性基底上制作的二维胶束隧穿器件;(f)柔性基底器件在不同弯曲程度下的电流-压力曲线。
通过对所得菱形二维自组装形貌的组分分布进行探究,发现二维自组装形貌中半导体的PPV嵌段被上下两层的绝缘外延嵌段包夹着,组成了一种“三明治”结构。这种结构使得半导体和绝缘组分在二维形貌中得到了很好的分离,通过分子设计可以控制绝缘层的厚度,进而可以有效地控制基于二维材料构筑的隧穿器件的垂直电导率,并以此制造出纳米级的压力传感器(图2a)。所制备隧穿器件的开关电流比大于104,连通态时电流密度高达6000 A cm-2,具有极高的灵敏度与良好的耐受性(图2b-d)。同时用这种方法制备的隧穿器件在弯曲时仍然表现了不错的性能(图2e、f)。实验结果表明这些二维胶束型材料在微电子或电子机械传感领域有着广阔的应用前景,如果借助嵌段共聚物分子外接功能链与羟基或金属离子之间进行相互作用,甚至可以通过衬底工程实现图案化的器件设计。
南方科技大学格拉布斯研究院研究助理教授韩亮以及物理系联培博士生凡华是该论文共同第一作者,南方科技大学化学系何凤副教授和物理系赵悦副教授为共同通讯作者。此外,南方科技大学物理系讲席教授、量子科学与工程研究院院长俞大鹏院士和北京大学新材料学院院长潘锋教授也对该论文有着重要贡献。本研究得到了国家自然科学基金委、深圳市科创委、深圳市诺贝尔奖科学家实验室项目、广东省催化重点实验室、广东省重点领域研发项目等部门的基金支持。该工作以Research article形式发表在CCS Chemistry,并在官网“Just Published”栏目上线。
文章详情:
Precisely Controlled Two-Dimensional Rhombic Copolymer Micelles for Sensitive Flexible Tunneling Devices
Liang Han , Hua Fan, Yulin Zhu , Meijing Wang , Feng Pan, Dapeng Yu, Yue Zhao * & Feng He*
Citation:CCS Chem. 2020, 2, 1399–1409


