-
[1]
Ren J, Shi X, Li X N, et al. Org. Lett., 2016, 18(16):3948~3951. doi: 10.1021/acs.orglett.6b01654
-
[2]
Zhu Y, Shao L D, Deng Z T, et al. J. Org. Chem., 2018, 83(17):10166~10174. doi: 10.1021/acs.joc.8b01424
-
[3]
Xu J, Shao L D, Li D S, et al. J. Am. Chem. Soc., 2014, 136(52):17962~17965. doi: 10.1021/ja5121343
-
[4]
Godula K, Sames D. Science, 2006, 312(5770):67~72. doi: 10.1126/science.1114731
-
[5]
He J, Wasa M, Chan K S L, et al. Chem. Rev., 2017, 117(13):8754~8786. doi: 10.1021/acs.chemrev.6b00622
-
[6]
Vargová D, Némethová L, Plevová K, et al. ACS Catal., 2019, 9(4):3104~3143. doi: 10.1021/acscatal.8b04357
-
[7]
Murahashi T, Kurosawa H. J. Organomet. Chem., 1999, 574(1):142~147. doi: 10.1016/S0022-328X(98)00934-6
-
[8]
Ciufolini M A, Qi H B, Browne M E. J. Org. Chem., 1988, 53(17):4149~4151. doi: 10.1021/jo00252a064
-
[9]
Muratake H, Natsume M. Tetrahed. Lett., 1997, 38(43):7581~7582. doi: 10.1016/S0040-4039(97)01787-5
-
[10]
Palucki M, Buchwald S L. J. Am. Chem. Soc., 1997, 119(45):11108~11109. doi: 10.1021/ja972593s
-
[11]
Svennebring A, Garg N, Nilsson P, et al. J. Org. Chem., 2005, 70(12):4720~4725. doi: 10.1021/jo0504619
-
[12]
Storgaard M, Dörwald F Z, Peschke B, et al. J. Org. Chem., 2009, 74(14):5032~5040. doi: 10.1021/jo900799y
-
[13]
Bélanger É, Cantin K, Messe O, et al. J. Am. Chem. Soc., 2007, 129(5):1034~1035. doi: 10.1021/ja067501q
-
[14]
Solé D, Vallverdú L, Solans X, et al. J. Am. Chem. Soc., 2003, 125(6):1587~1594. doi: 10.1021/ja029114w
-
[15]
Huang Z, Chen Z L, Lim L H, et al. Angew. Chem. Int. Ed., 2013, 52(22):5807~5812. doi: 10.1002/anie.201300481
-
[16]
hman J, Wolfe J P, Troustman M V, et al. J. Am. Chem. Soc., 1998, 120(8):1918~1919. doi: 10.1021/ja973794z
-
[17]
Kawatsura M, Hartwig J F. J. Am. Chem. Soc., 1999, 121(7):1473~1478. doi: 10.1021/ja983378u
-
[18]
Fox J M, Huang X H, Chieffi A, et al. J. Am. Chem. Soc., 2000, 122(7):1360~1370. doi: 10.1021/ja993912d
-
[19]
Ehrentraut A, Zapf A, Beller M. Adv. Synth. Catal., 2002, 344(2):209~217.
-
[20]
Liao X, Weng Z, Hartwig J F. J. Am. Chem. Soc., 2008, 130(1):195~200. doi: 10.1021/ja074453g
-
[21]
Viciu M S, Germaneau R F, Nolan S P. Org. Lett., 2002, 4(23):4053~4056. doi: 10.1021/ol026745m
-
[22]
Hamada T, Chieffi A, Åhman J, et al. J. Am. Chem. Soc., 2002, 124(7):1261~1268. doi: 10.1021/ja011122+
-
[23]
Liao X, Stanley L M, Hartwig J F. J. Am. Chem. Soc., 2011, 133(7):2088~2091. doi: 10.1021/ja110215b
-
[24]
Han C, Kim E H, Colby D A. J. Am. Chem. Soc., 2011, 133(15):5802~5805. doi: 10.1021/ja202213f
-
[25]
Jiao Z W, Beiger J J, Jin Y S, et al. J. Am. Chem. Soc., 2016, 138(49):15980~15986. doi: 10.1021/jacs.6b09580
-
[26]
Shaughnessy K H, Hamann B C, Hartwig J F. J. Org. Chem., 1998, 63(19):6546~6553. doi: 10.1021/jo980611y
-
[27]
Durbin M J, Willis M C. Org. Lett., 2008, 10(7):1413~1415. doi: 10.1021/ol800141t
-
[28]
Altman R A, Hyde A M, Huang X H, et al. J. Am. Chem. Soc., 2008, 130(29):9613~9620. doi: 10.1021/ja803179s
-
[29]
Taylor A M, Altman R A, Buchwald S L. J. Am. Chem. Soc., 2009, 131(29):9900~9901. doi: 10.1021/ja903880q
-
[30]
Jin Y S, Chen M, Ge S Z, et al. Org. Lett., 2017, 19(6):1390~1393. doi: 10.1021/acs.orglett.7b00294
-
[31]
Xu Y, Su T S, Huang Z X, et al. Angew. Chem. Int. Ed., 2016, 55(7):2559~2563. doi: 10.1002/anie.201510638
-
[32]
Liu R R, Li B L, Lu J, et al. J. Am. Chem. Soc., 2016, 138(16):5198~5201. doi: 10.1021/jacs.6b01214
-
[33]
Zhu C D, Wang D Y, Sun W Y, et al. J. Am. Chem. Soc., 2017, 139(46):16486~16489. doi: 10.1021/jacs.7b10365
-
[34]
Huang X, Oh W R J J, Zhou J S. Angew. Chem. Int. Ed., 2018, 57(26):7673~7677. doi: 10.1002/anie.201804318
-
[35]
Wang M, Chen J, Chen Z J, et al. Angew. Chem. Int. Ed., 2018, 57(10):2707~2711. doi: 10.1002/anie.201711845
-
[36]
Liu X, Hartwig J F. Org. Lett., 2003, 5(11):1915~1918. doi: 10.1021/ol034570q
-
[37]
Jiang L, Weist S, Jansat S. Org. Lett., 2009, 11(7):1543~1546. doi: 10.1021/ol900131q
-
[38]
Fernández-Nieto F, Mas Roselló J, Lenoir S, et al. Org. Lett., 2015, 17(15):3838~3841. doi: 10.1021/acs.orglett.5b01803
-
[39]
Hyde A M, Buchwald S L. Angew. Chem. Int. Ed., 2008, 47(1):177~180. doi: 10.1002/anie.200704529
-
[40]
Zhao Y H, Zhou Y Y, Liang L L, et al. Org. Lett., 2009, 11(3):555~558. doi: 10.1021/ol802608r
-
[41]
Johnson T, Pultar F, Menke F, et al. Org. Lett., 2016, 18(24):6488~6491. doi: 10.1021/acs.orglett.6b03394
-
[42]
Hou W Y, Wu Y K. Org. Lett., 2017, 19(5):1220~1223. doi: 10.1021/acs.orglett.7b00268
-
[43]
Yang Y C, Lin Y C, Wu Y K. Org. Lett., 2019, 21(23):9286~9290. doi: 10.1021/acs.orglett.9b03071
-
[44]
Semmelhack M F, Stauffer R D, Rogerson T D. Tetrahed. Lett., 1973, 14(45):4519~4522. doi: 10.1016/S0040-4039(01)87265-8
-
[45]
Spielvogel D J, Buchwald S L. J. Am. Chem. Soc., 2002, 124(14):3500~3501. doi: 10.1021/ja017545t
-
[46]
Chen G S, Kwong F Y, Chan H O, et al. Chem. Commun., 2006, (13):1413~1415. doi: 10.1039/b601691j
-
[47]
Ge S, Hartwig J F. J. Am. Chem. Soc., 2011, 133(41):16330~16333. doi: 10.1021/ja2082087
-
[48]
Josep C, Jackson E P, Martin R. Angew. Chem. Int. Ed., 2015, 54(13):4075~4078. doi: 10.1002/anie.201412051
-
[49]
Ghosh A, Walker J A, Arkady A, et al. ACS Catal., 2016, 6(4):2673~2680. doi: 10.1021/acscatal.6b00365
-
[50]
Harvey J S, Simonovich S P, Jamison C R, et al. J. Am. Chem. Soc., 2011, 133(35):13782~13785. doi: 10.1021/ja206050b
-
[51]
Liu T, Feng J J, Chen C, et al. Org. Lett., 2019, 21(12):4505~4509. doi: 10.1021/acs.orglett.9b01373
-
[52]
Baran P S, Richter J M. J. Am. Chem. Soc., 2004, 126(24):7450~7451. doi: 10.1021/ja047874w
-
[53]
Baran P S, Richter J M. J. Am. Chem. Soc., 2005, 127(44):15394~15396. doi: 10.1021/ja056171r
-
[54]
Baran P S, Maimone T J, Richter J M. Nature, 2007, 446(7134):404~408. doi: 10.1038/nature05569
-
[55]
Wu H R, Huang H Y, Ren C L, et al. Chem. Eur. J., 2015, 21(47):16744~16748. doi: 10.1002/chem.201502519
-
[56]
Guo J, Dong S X, Zhang Y L, et al. Angew. Chem. Int. Ed., 2013, 52(39):10245~10249. doi: 10.1002/anie.201303602
-
[57]
Ueda N, Tokuyama T, Sakan T. Bull. Chem. Soc. Jpn., 1966, 39(9):2012~2014. doi: 10.1246/bcsj.39.2012
-
[58]
Stewart J D, Fields S C, Kochhar K S, et al. J. Org. Chem., 1987, 52(10):2110~2113. doi: 10.1021/jo00386a045
-
[59]
Picazo E, Anthony S M, Giroud M, et al. J. Am. Chem. Soc., 2018, 140(24):7605~7610. doi: 10.1021/jacs.8b02875
-
[60]
Semmelhack M F, Bargar T M. J. Org. Chem., 1977, 42(8):1481~1482. doi: 10.1021/jo00428a056
-
[61]
Liang K, Li N, Zhang Y, et al. Chem. Sci., 2019, 10(10):3049~3053. doi: 10.1039/C8SC05170D
-
[62]
Shirakawa S, Koga K, Takuda T, et al. Angew. Chem. Int. Ed., 2014, 53(24):6220~6223. doi: 10.1002/anie.201403046
-
[63]
Sattar M, Rathore V, Prasad C D, et al. Chem. Asian J., 2017, 12(7):734~743. doi: 10.1002/asia.201601647
-
[64]
Rao X F, Li N K, Bai H, et al. Angew. Chem. Int. Ed., 2018, 57(38):12328~12332. doi: 10.1002/anie.201807302




 下载:
下载:













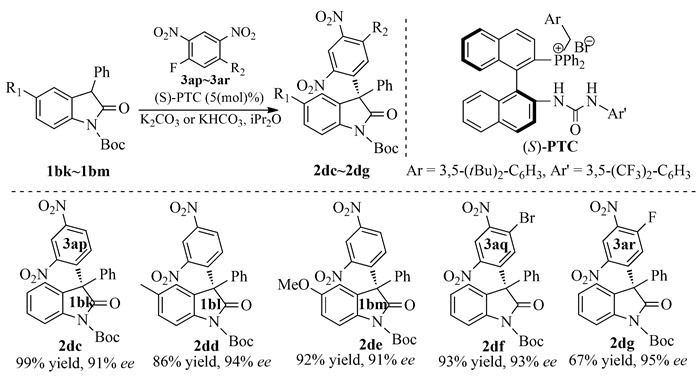

 下载:
下载:














