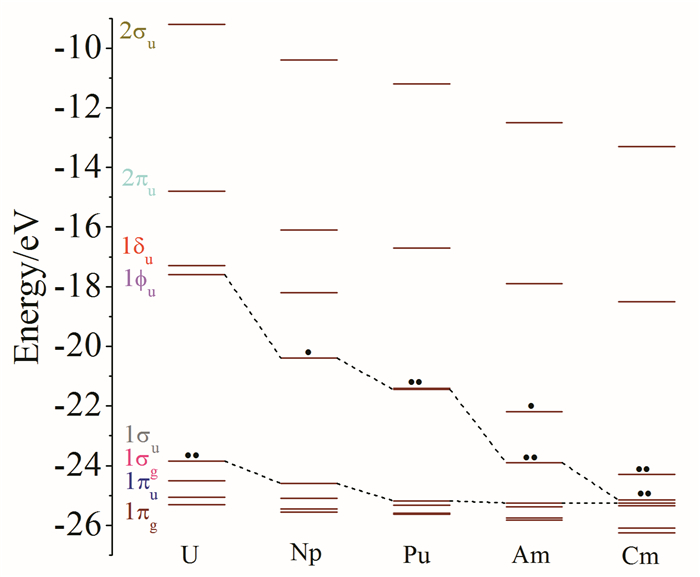
图 1.
(AnO2)2+的轨道能级图
Figure 1.
Scheme for the molecular orbital diagram of free [AnO2]2+ (An=U-Cm)

核能作为一种能量密度高、洁净、低碳的理想能源备受人们关注。随着世界核电反应堆运营年数和新建核电机组投产数量的增加,核燃料后处理愈发成为焦点和热点。辐射后的核燃料(乏燃料)元件含有极强的放射性和尚未被充分利用的铀、钚资源,如果不经处理,直接地质贮存,不仅浪费了有限的核资源,而且对地球环境会带来长期威胁。因此,如何安全、有效、经济地进行乏燃料后处理已成为核工业尤其是核能能否可持续发展的重大问题之一。在先进核燃料循环中,目前国际上普遍优先采用PUREX(Plutonium Uranium Reduction Extraction)流程联合高放废液(High level liquid waste,HLLW)“分离-嬗变”工艺处理乏燃料。首先通过铀钚萃取法分离、回收铀和钚,充分地实现铀和钚的再循环,从而极大地提高核燃料资源的利用率。然而经PUREX流程后产生的高放废液中包含着大量长寿命、具有极强放射性的次锕系元素(MAs),如镅、锔,对人类健康和生态环境有着直接潜在危害,需要对其进行嬗变处理来最大限度地减小高放废物的体积及其放射性毒性,实现核废物的最小化,防止对人类和环境的危害。因为高放废液中还含有大量的具有较大中子俘获面积的镧系元素而降低了嬗变效率,所以需要将次锕系元素从镧系元素中分离出来。由于锕系与镧系元素具有非常相似的物理化学性质,因此镧锕分离成为高放废液处理中最具挑战的研究课题之一。另外,锕系元素一般具有很强的放射毒性,特别是次锕系元素,开展实验研究有较大困难和挑战,因此从理论上设计镧锕分离配体是非常必要的[1~4]。
当前实验研究中萃取剂的设计与选择主要基于经验的软硬酸碱理论,即,锕系元素的5f和6d轨道比镧系元素的4f轨道径向分布伸展更长,使得锕系离子可以与配体形成微弱的共价键,因此利用萃取剂对于不同元素之间的配位差异对锕系元素进行选择性分离,其关键在于选取高效的萃取剂。例如,利用含磷萃取剂从水合物中实现三价锕/镧系元素的分离过程(TALSPEAK)。这类配体由于用硫原子部分或全部取代了氧原子,从而降低了配体的相对硬度,使配体对锕系元素和镧系元素萃取能力的差异增大。近年来,一些包含氮、氧、硫、磷等配位原子的配体(BTP、BTBP、BTPhen、Cyanex系列等)被开发出来,并被用于开展镧锕系元素分离的研究,而主要的相关工作仍停留于实验室级别的测试,且很多萃取体系各有优缺点,距离实现核工业的实际应用目标存在一定差距。另一方面,开发新型的、更高效的配体是克服镧锕分离这一难题的关键所在,也是国内外从事相关研究的学者们关注的焦点。在众多候选配体萃取剂中,大多数不能在低pH的环境中存在。然而,有一类可以容忍高酸度的配体,即冠醚及其衍生物,同时它们对镅、锔的分离因子超过铕的100倍[6~11]。
笔者研究组在近年来开展了一系列对锕系化合物电子结构和吸收光谱的理论研究工作,并取得了一些初步的结果[12~21]。本文将结合这些结果介绍我们在锕酰冠醚配合物成键性质和化学选择性理论研究方面的进展。
众所周知,锕酰是直线型结构,其所属对称点群为D∞h,在此对称性下,5f轨道分裂成fσu轨道和三个双重简并的fπu,fδu和fϕu轨道;6d分裂成dσg轨道以及两个双重简并的dπg和dδg轨道。从纯离子键图像上来看,所有的价电子来源于轴向O2-,An因失去6个价电子,其氧化态为+6。O2-中电子全充满的2p价轨道与对称性匹配的An5f和6d轨道相互作用而能量下降,形成6个成键轨道(3σu、3σg、2πu和1πg),对应于An≡O三重键,以及相应的6个反键空轨道。An的5fδu、5fϕu和6dδg轨道,由于与轴向O的价层轨道对称性不匹配,无法参与An-O成键,形成非键空轨道1δu、1ϕu和1δg; 而当赤道平面有路易斯碱配体配位时,它们作为电子给体可以与这些非键轨道形成弱的σ和π相互作用[22]。图 1展示了锕酰离子AnO22+中,从U到Cm轨道能级的变化趋势。由于An-6d和An-7s轨道能级较高,且在此系列化合物中基本不变,故未在图中标注。随着原子核电荷数的增加,配体向金属的电荷转移(LMCT)以及显著的组态混合将导致Cm的5fπu*轨道上占据更多的电子,这将导致从U到Cm,锕酰的电子结构呈现以下趋势:[UVI(≡Oyl2-)2]2+→ [PuVI(≡Oyl2-)2]2+→[CmIV(=Oyl1-)2]2+。An上有效电荷数的减少将削弱离子相互作用,所以Pu≡Oyl到Am≡Oyl再到Cm≡Oyl的“锕系收缩”现象不明显。An-5f轨道上布居的电子数越来越多,表明Am和Cm的氧化态很难保持形式上的+6价[23, 24]。
我们采用密度泛函理论方法,选取铀酰(UO22+)、镎酰(NpO22+)、钚酰(PuO22+)、镅酰(AmO22+)、锔酰(CmO22+)离子与两种冠醚和两种硫代冠醚的相互作用,分析形成的配合物的几何结构和电子结构。我们的研究考虑了下列几种可能情况:(1)单环12C4和15C5配体配位锕酰离子;(2)双环12C4和15C5配体配位铀酰离子;(3)单环和双环的12TC4和15TC5配体配位锕酰离子。对于单环冠醚及硫代冠醚与锕酰离子AnO22+的配位模式,主要考虑了“侧配位”和“嵌入式”两种异构体;对于双环结构,考虑了“嵌入式”结构和“侧配位夹心式”两种异构体。
研究表明,在所有[AnO2(15C5)]2+(An=U-Cm)基态稳定结构中,冠醚均呈现嵌入式结构,通过冠醚O原子以五齿的方式与锕离子在赤道面配位,这与之前报道的锕酰配位模式一致(见图 2)[25~33]。单环15C5配合物中,配合物呈C2对称性,轴向An-Oyl键长伸长。以[UO2(15C5)]2+为例,U-Oyl键长约为1.784Å,稍长于EXAFS报道的[UO2]2+中的U-Oyl键长,1.77Å[34]。赤道平面上U-O15C5平均键长为2.411Å。通过计算[AnO2(15C5)]2+(An=U-Cm)各物种的稳定化能ΔE>200kcal/mol,我们发现溶剂化效应使得这些配合物更趋稳定。此外,在这些[AnO2(15C5)]2+(An=U-Cm)中,随着锕系原子的原子序数增大,An-O15C5键长小幅度减小,这主要与锕系原子的轨道收缩有关[17]。
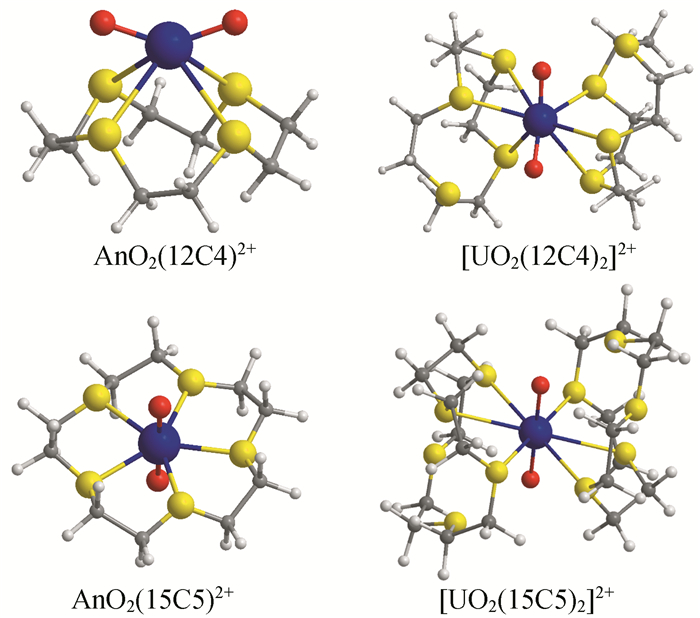
然而,图 2中单环12C4配位时冠醚呈现四齿侧配位,具有C2对称性。由于锕酰离子和配位氧原子的相互作用较强,破坏了锕酰离子的直线型结构,锕酰离子键长增长,参与成键的5f轨道成分减小。例如,铀酰离子从线性结构变为角度是133°的折线结构,虽然它的U-Oyl键长是典型的铀酰离子键长但明显增长,为1.800Å,平均U-O12C4键长分别为2.578Å。这是因为相较于15C5,12C4冠醚腔体空穴较小,不能容纳锕酰离子嵌入环中,从而形成侧配位结构[15]。
双环冠醚配体配位时,冠醚均以侧配位的方式与UO22+离子形成夹心结构(见图 2)。在[UO2(15C5)2]2+结构中,形成具有Ci对称性的几何结构,两个冠醚配体各提供3个O原子,以六齿的方式与U原子配位。6个配位的O15C5原子共面但不垂直于铀酰离子。配位键U-O15C5键长范围在2.566~2.822 Å之间。除此之外的U-O15C5键长大于3Å,超出了U-O15C5配位键键长范围。与单个冠醚形成的嵌入式结构配合物相比,U-O15C5键长明显变长,同时U≡Oyl键长明显缩短,为1.744Å。这一结构合理地解释了实验报道的相关化合物的光谱性质。在[UO2(12C4)2]2+结构中,每个12C4通过O12C4原子以三齿的方式与U原子配位。我们的研究表明,铀酰离子铀酰赤道平面最佳配位数为6[12, 21]。
锕系元素配位化学的理论基础主要基于软硬酸碱理论,利用配体与锕系金属之间的共价性成键的差异来实现最终的分离过程。从这方面来说,相比于冠醚中的硬碱氧原子,硫代冠醚中的硫原子是软碱,S-3p轨道径向分布更延展,可以有效地与锕系元素的5f轨道重叠。理论研究表明,硫代冠醚与锕酰离子AnO22+形成配位化合物的几何结构与冠醚与锕酰离子AnO22+的配位化合物结构类似,即与单环15TC5硫代冠醚形成“嵌入结构”,与单环12T4硫代冠醚形成侧配位结构,与双环15TC5和12TC4均形成侧配位夹心结构。与冠醚[AnO2(15C5)]2+(An=U-Cm)对比,锕系离子与硫原子之间的成键强度较弱,An-S键长范围在2.91~3.08 Å之间,远远大于An-O15C5的键长(2.41~2.59Å)。同时,在硫代冠醚[AnO2(15TC5)]2+(An=U-Cm)结构中,An≡Oyl键的键长较短,这说明锕酰离子受配体场影响较小[16]。
表 1显示了从U到Pu化合物中An-OL和An≡Oyl的平均键长RAn-OL和RAn≡Oyl变化趋势。在这些冠醚化合物中,RAn≡Oyl均呈现“抛物线”形状,即从U到Am化合物,RAn≡O逐渐变小,然后在Am和Cm化合物中变大。这一现象在其他的六价锕酰化合物中也有体现。而An-OL的键长在U到Cm中基本不变,在Cm中变长,其原因为静电相互作用变弱。这一规律在其他的锕系化合物中也有体现[35, 36]。
 下载:
导出CSV
下载:
导出CSV
| Species | [(AnO2)(L)]2+ | |||||||||||
| U | Np | Pu | Am | Cm | Species | U | Np | Pu | Am | Cm | ||
| 12C4 | An-Oe | 2.578 | 2.603 | 2.610 | 2.627 | 2.624 | 12TC4 | An-S | 3.000 | 3.009 | 3.103 | 3.464 |
| An-Oyl | 1.800 | 1.772 | 1.768 | 1.765 | 1.775 | An-Oyl | 1.796 | 1.787 | 1.775 | 1.766 | ||
| 15C5 | An-Oe | 2.401 | 2.396 | 2.395 | 2.392 | 2.380 | 15TC5 | An-S | 2.931 | 2.921 | 2.915 | 2.906 |
| An-Oyl | 1.792 | 1.771 | 1.762 | 1.755 | 1.763 | An-Oyl | 1.789 | 1.772 | 1.768 | 1.765 | ||
| Species | [(UO2)(L)2]2+ | |||||||||||
| 12C4 | 15C5 | |||||||||||
| An-Oyl | 1.755 | 1.772 | ||||||||||
| An-Oe | 2.732 | 2.732 | 3.071 | 3.071 | 2.564 | 2.699 | 2.822 | 3.503 | 3.531 | |||
通过轨道的径向分布函数图可以进一步理解上述的键长变化。图 3展示了An6+的5f和6d轨道的径向分布函数图,为了定性地讨论轨道重叠,将OL和SL放在距离An6+ 250pm和300pm的位置,并绘出了O-2p和S-3p轨道的径向分布函数图。随着核电荷数的增加,An6+的价层轨道均逐渐收缩。其中An-6d轨道与O-2p轨道可以有效地重叠,故在成键过程中起主要作用,但影响其成键效果的因素是它的能量较高。随着轨道的收缩,An-6d轨道与O-2p轨道的有效重叠也逐渐变小,这将导致由于反馈键产生的An-6d轨道上的电子占据数逐渐变少。An-5f轨道因为过于收缩,它与O-2p轨道的重叠很小,而且从U到Cm逐渐变小。这也使得An-5f轨道上的电子越来越难以失去,故Cm很难保持+6价。类似地,An-6d轨道与SL的3p轨道有效重叠,在成键中占主要部分,而An-5f轨道与SL的3p轨道的重叠明显很微弱,且随着核电荷数增加,An价层轨道与S-3p轨道的重叠逐渐减小。Bursten等[37, 38]将5f轨道基本不受配体影响,而6d轨道主要参与和配体的相互作用这一成键规律定义为“FEUDAL”(f’s essentially unaffected,d accommodates ligands)模型。
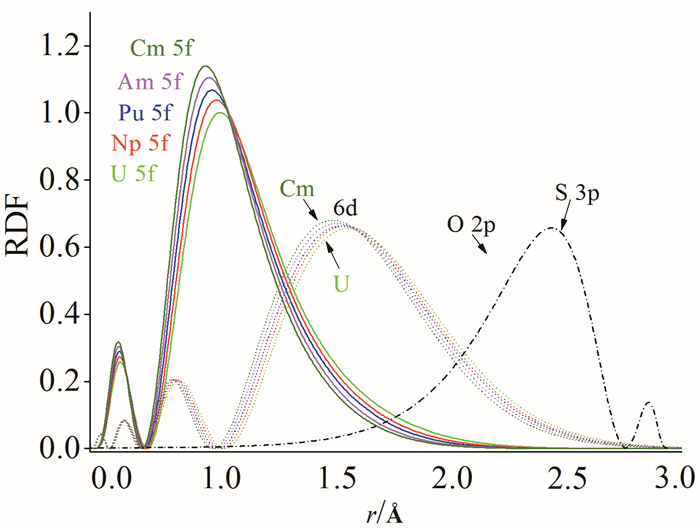
O-2p(黑色实线)和S-3p(黑色虚线)的径向轨道密度的重叠分别以弱结合的O与An(典型距离为2.4Å)和弱结合的S与An(3.0Å)为中心
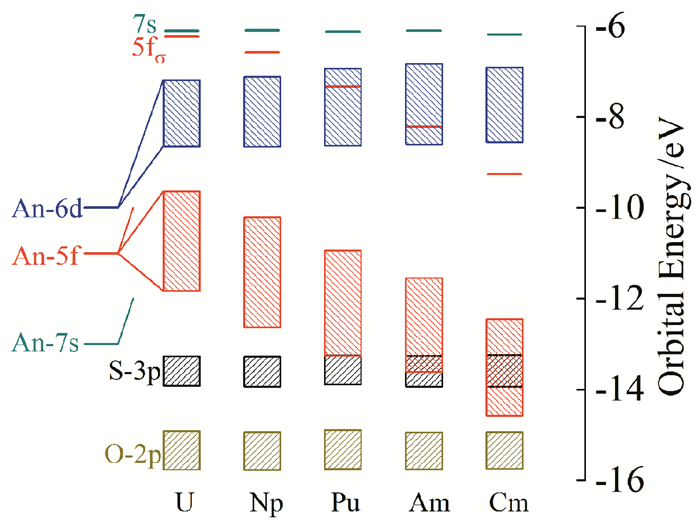
在上述讨论中,我们注意到锕酰离子AnO22+的An-5f型轨道的能级顺序为δu ≈ ϕu < πu < < δu,其中πu和δu为反键轨道。从UO22+(f0)到CmO22+(f4),随着5f电子的增多,一般认为这些电子将依次占据非键轨道5fδu和5fϕu。但是这一图像过于简单,并没有考虑到从U到Cm,5f轨道能量将不断降低以及径向分布变得越来越收缩。随着An-5f型轨道的能量逐渐降低,将会和配体轨道接近从而增加轨道之间的混合。此时LMCT会非常显著,电子密度将从配体成键轨道转移到反键轨道5fπu,进而削弱An≡Oyl键的强度,使得An的氧化态降低,这也是CmO22+不能稳定存在的内在原因,如图 4所示。
在[AnO2 (L)n]2+ (An=U-Cm;L=12C4,15C5;n=1,2)化合物中,AnO22+和冠醚两个碎片之间的相互作用主要为库伦作用,很多碎片轨道变化不显著,仅仅是由于静电相互作用造成能级改变。以[UO2(15C5)2]2+为例(如图 5所示),[UO2(15C5)2]2+的前线分子轨道按能量从低到高分别为UO22+中的U-5f6d/Oyl-2p的成键轨道,然后是15C5中O-2p形成的非键轨道。HOMO-1(52au)、HOMO-3(51au)、HOMO-4(50au)和HOMO-7(49au)对应于UO22+和冠醚之间配位键,它们分别由是U-6dδ和U-5fφ与15C5的HOMO轨道相互作用产生,对应于3个反馈σ键。54au和55au主要来自U-5fδu轨道,(UO2)2+中原本简并的U-5fϕu轨道在Ci对称性分裂为两个au,其中56au与来自15C5碎片的HOMO轨道作用形成成键轨道50au和反键轨道56au,而5fϕ中的另一个au轨道在[UO2(15C5)2]2+中依然为非键轨道(53au)。从Np到Cm化合物,将分别有1~4个单电子占据53au、54au、55au和56au轨道。
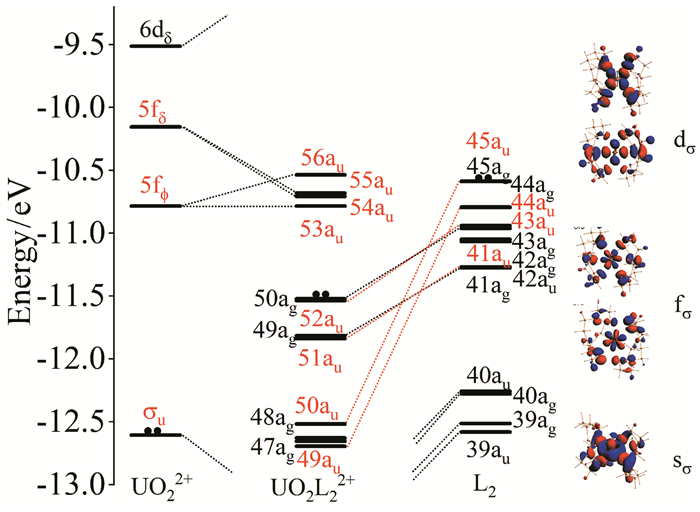
根据ETS-NOCV分析,UO22+和(15C5)2单元相互作用的片段之间的代表性变形密度(isovalue=0.005)等高线(红色区域表示电子流出,蓝色表示电子流入该区域)
在本文中我们尝试从锕酰冠醚配合物的几何结构和电子结构的理论研究出发,对锕酰冠醚配合物成键规律进行综述。根据我们的计算,获得以下规律性结果:(1)几何结构方面,在[AnO2(L)n]2+ (An=U-Cm;L=12C4,15C5,n=1,2)系列化合物中,锕酰中的An≡Oyl化学键较强,而An-OL配位键较弱;An-OL属于典型的配位键,受溶剂环境影响较大;An≡Oyl键属于共价键,在U到Am的化合物中,键长逐渐变短,符合“锕系收缩”的规律,但在Am之后An≡Oyl键长显著增长,An≡Oyl共价性明显变弱。(2)电子结构方面,在[AnO2(L)n]2+中,AnO22+离子与冠醚配体之间相互作用主要为库仑作用,AnO22+轨道能级变化不显著。从U到Cm的化合物中,5f轨道的逐渐收缩导致与Oyl-2p的重叠削弱,而且5f轨道能量逐渐降低和Oyl-2p轨道近简并,这使得5f上的电子难以失去,导致Am和Cm的氧化态降低。而且,O-2p和S-2p型轨道的能带基本保持不变,LMCT变得显著,锕酰与配体的作用变弱,An-OL或An-S键长增长。(3)化学成键方面,在锕酰配合物中,6d轨道主要参与化学成键,5f轨道起到调节锕系元素氧化态的作用,进而影响锕系离子和配体之间的静电相互作用。这一规律可以扩展到整个锕系化合物中,用于预测后锕系元素的氧化态,以及预测镧锕分离的有效性,为镧/锕分离萃取剂的设计提供思路。
顾忠茂, 柴之芳.化学进展, 2011, 23(7): 1263~1271. http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201107003.htm
韦悦周.化学进展, 2011, 23(07): 1272~1288. http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201107004.htm
王祥云, 陈涛, 刘春立.化学进展, 2011, 23(07): 1400~1410. http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201107018.htm
Vidaud C, Bourgeois D, Meyer D. Chem. Res. Toxicol., 2012, 25(6): 1161~1175. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM22458510
李振祥, 刘世昌, 倪嘉缵, 等.化学学报, 1990, 48(10): 1006~1010. http://www.cnki.com.cn/Article/CJFDTotal-HXXB199010011.htm
Gokel G W, Leevy W M, Weber M E. Chem. Rev., 2004, 104: 2723~2750. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM15137805
Pedersen C J, Frensdor H K. Angew. Chem. Int. Ed., 1972, 11: 16~25. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM4622977
Cooper T E, Carl D R, Oomens J, et al. J. Phys. Chem. A 2011, 115: 5408~5422. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM21561140
Servaes K, De Houwer S, Görller-Walrand C, et al. Phys. Chem. Chem. Phys., 2004, 6: 2946~2950. https://www.researchgate.net/publication/230687715_Spectroscopic_properties_of_uranyl_chloride_complexes_in_non-aqueous_solvents
Shamov G A, Schreckenbach G, Martin R L, et al. Inorg. Chem., 2008, 47: 1465~1475. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM18225857
Gong Y, Gibson J K. Inorg. Chem., 2014, 53: 5839~5844. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM24828467
Hu S X, Gibson J K, Li W L, et al. Chem. Commun., 2016, 52: 12761~12764. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM23950730
Hu S X, Chen M, Ao B. Phys. Chem. Chem. Phys., 2018, 20: 23856~23863.
Hu S X, Liu H T, Liu J J, et al. ACS Omega, 2018, 3(10): 13902~13912.
Jian J W, Hu S X, Li W L, et al. Inorg. Chem., 2018, 57(7): 4125~4134.
Hu S X, Liu J J, Gibson J K, et al. Inorg. Chem., 2018, 57(5): 2899~2907. https://www.researchgate.net/publication/323269082_Periodic_Trends_in_Actinyl_Thio-Crown_Ether_Complexes
Hu S X, Li L W, Dong L, et al. Dalton Transac., 2017, 46(36): 12354~12363.
Hu S X, Gibson J K, Li W L, et al. Chem. Commun., 2016, 52(86): 12761~12764.
Hu S X, Lu E, Liddle S T. Dalton Transac., 2019, 48(34): 12867~12879.
Qin J, Zhang P, Pu Z, et al. J. Phys. Chem. A, 2019, 123(32): 6958~6969.
Hu S X, Jian J, Li J, et al. Inorg. Chem., 2019, 58(15): 10148~10159.
Pepper M, Bursten B E. Chem. Rev., 1991, 91: 719~741. https://www.researchgate.net/publication/231245256_ChemInform_Abstract_The_Electronic_Structure_of_Actinide-Containing_Molecules_A_Challenge_to_Applied_Quantum_Chemistry
Kovacs A, Konings R J M, Gibson J K, et al. Chem. Rev. 2015, 115(4): 1725~1759. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM25679237
Marcalo J, Gibson J K. J. Phys. Chem. A, 2009, 113(45): 12599~12606. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM19725530
Pyykkö P, Li J, Runeberg N. J. Phys. Chem., 1994, 98(18): 4809~4813. https://www.researchgate.net/publication/232326438_Predicted_ligand_dependence_of_the_AuIAuI_attraction_in_XAuPH32
Kovácsa A, Konings R J M. J. Mol. Struct., 2004, 684(1/3): 35~42.
Liu J B. Chen G P. Huang W, et al. Dalton Transac., 2017, 46: 2542~2550.
Su J, Dau P D, Liu H T, et al. J. Chem. Phys., 2015, 142(13): 134308.
Fujii T, Uehara A.; Kitatsuji Y, et al. J. Radioanal. Nucl. Chem., 2015, 303(1): 1015~1020.
Lan J H, Wang C Z, Wu Q Y, et al. J. Phys. Chem. A, 2015, 119(34): 9178~9188.
Shamov G A. J. Am. Chem. Soc., 2011, 133(12): 4316~4329. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM21381733
Wang Y L, Liu Z Y, Li Y X, et al. J. Am. Chem. Soc., 2015, 137(19): 6144~6147.
Neuefeind J, Soderholm L, Skanthakumar S. J. Phys. Chem. A, 2004, 108: 2733~2739. https://www.researchgate.net/publication/231638180_Experimental_Coordination_Environment_of_UranylVI_in_Aqueous_Solution
Kovács A, Pogany P, Konings R. Inorg. Chem., 2012, 51(8): 4841~4849. https://www.researchgate.net/publication/223984256_Theoretical_Study_of_Bond_Distances_and_Dissociation_Energies_of_Actinide_Oxides_AnO_and_AnO2
Sokolova M N, Fedosseev A M, Andreev G B, et al. Inorg. Chem., 2009, 48(19): 9185~9190. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM19725546
Burns C J, Bursten B E. Comment. Inorg. Chem., 1989, 9(2): 61~93. https://www.researchgate.net/publication/233202549_Covalency_in_f-Element_Organometallic_Complexes_Theory_and_Experiment
Bursten B E, Palmer E J, Sonnenberg J L. Special Publication-Royal Society of Chemistry, 2006, 305(1): 157~162. https://www.ingentaconnect.com/content/rsoc/02606291/2006/00000305/00000001/art00043

图 1 (AnO2)2+的轨道能级图
Figure 1 Scheme for the molecular orbital diagram of free [AnO2]2+ (An=U-Cm)

图 2 [(AnO2)(L)]2+、[(UO2)(12C4)2]2+和[(UO2)(15C5)2]2+ (An=U-Cm; L=12C4, 15C5, 12TC4 and 15TC5)的几何构型
Figure 2 Optimized geometries of [AnO2(L)]2+, [(UO2)(12C4)2]2+ and [(UO2)(15C5)2]2+(An=U-Cm; L=12C4, 15C5, 12TC4 and 15TC5). Selected parameters are listed in Table 1

图 3 An6+的5f、6d以及O2-的2p轨道和S2-的3p轨道径向分布函数图[16]
Figure 3 Atomic valence-orbital radial-densities D(r)=r2R(r)2 of 5f (solid line) and 6d (dotted line) orbitals of the atomic ions U6+ through Cm6+ from B3LYP density functional calculations[16]
O-2p(黑色实线)和S-3p(黑色虚线)的径向轨道密度的重叠分别以弱结合的O与An(典型距离为2.4Å)和弱结合的S与An(3.0Å)为中心

图 4 [AnO2(L)]2+ (An=U-Cm; L=15C5, 15TC5)的分子轨道能级图
Figure 4 Scalar relativistic molecular orbital energy levels of free [AnO2(L)]2+ (An=U-Cm; L=15C5, 15TC5)

图 5 [(UO2)(15C5)2]2+的轨道相互作用图
Figure 5 MO energy levels of [(UO2)(15C5)2]2+ Contours of representative deformation densities (isovalue=0.005) between the interacting fragments of UO22+ and (15C5)2 unit from ETS-NOCV analysis, describing the density inflow (blue) and outflow (red)
根据ETS-NOCV分析,UO22+和(15C5)2单元相互作用的片段之间的代表性变形密度(isovalue=0.005)等高线(红色区域表示电子流出,蓝色表示电子流入该区域)
表 1 在PBE方法和TZ2P基组下,结构优化后的结构数据(Å)
Table 1. Selected average bond length (Å) of An-Oyl and An-Oe for the ground state [AnO2(L)]2+, [(UO2)(12C4)2]2+and[(UO2)(15C5)2]2+ (An=U - Cm; L=12C4, 15C5, 12TC4 and 15TC5) at PBE/TZ2P level
| Species | [(AnO2)(L)]2+ | |||||||||||
| U | Np | Pu | Am | Cm | Species | U | Np | Pu | Am | Cm | ||
| 12C4 | An-Oe | 2.578 | 2.603 | 2.610 | 2.627 | 2.624 | 12TC4 | An-S | 3.000 | 3.009 | 3.103 | 3.464 |
| An-Oyl | 1.800 | 1.772 | 1.768 | 1.765 | 1.775 | An-Oyl | 1.796 | 1.787 | 1.775 | 1.766 | ||
| 15C5 | An-Oe | 2.401 | 2.396 | 2.395 | 2.392 | 2.380 | 15TC5 | An-S | 2.931 | 2.921 | 2.915 | 2.906 |
| An-Oyl | 1.792 | 1.771 | 1.762 | 1.755 | 1.763 | An-Oyl | 1.789 | 1.772 | 1.768 | 1.765 | ||
| Species | [(UO2)(L)2]2+ | |||||||||||
| 12C4 | 15C5 | |||||||||||
| An-Oyl | 1.755 | 1.772 | ||||||||||
| An-Oe | 2.732 | 2.732 | 3.071 | 3.071 | 2.564 | 2.699 | 2.822 | 3.503 | 3.531 | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们