表 1
各种多元硫化物电解质的离子电导率(S·cm-1)
Table 1.
Ionic conductivity of various polysulfide solid electrolytes (S·cm-1)

锂离子电池在1990年被索尼公司[1]第一次商业化后,因具有能量密度高和质量轻等优点,正逐步取代铅酸和镍氢电池,成为最常用的储能技术之一[2, 3]。现阶段,商业化锂离子电池主要采用液态电解质,复杂的固/液界面反应、较差的热稳定性、漏液以及有毒等问题限制了其进一步的发展。固体电解质多为陶瓷和非晶态玻璃体等材料,其在能量密度、安全性以及稳定性等方面较液态电解质具有无法比拟的优点。无机固态电解质主要包括氧化物和硫化物体系,其中硫化物体系的离子电导率比氧化物体系更高。这是因为相比于O2-,S2-的电负性更弱,导致其对Li+的约束力更小;其次,S2-的半径更大,使Li+离子迁移通道更宽;而且,S2-有更高的极化能力[4, 5]。据报道,硫化物电解质Li10GeP2S12在室温下的电导率高达1.2×10-2S·cm-1,接近液态电解质[6]。目前,硫化物电解质还存在原材料昂贵、室温下电导率较低、热稳定性差和易吸水生成剧毒H2S气体等问题。已报道的硫化物电解质主要分为Li2S-P2S5、Li2S-SiS2和thio-LISICON类固溶体,本文主要介绍了Li2S-P2S5体系电解质的合成、掺杂改性、表征方法以及电极/固态电解质之间的固-固界面稳定性的研究进展。
熔体淬火法是制备无机固态电解质的传统方法,典型的制备过程为:首先将原料按一定比例混合并密封在碳涂层的石英管中,置于熔炉中进行高温熔融反应后慢慢冷却至室温或在冰水中淬火得到最终产物。虽然熔体淬火操作简单,但反应条件不好控制易导致杂质相产生,且工作温度高,安全性低(P2S5有较高蒸气压,熔化过程必须在一个密封的石英管中进行)。采用熔体淬火法制备的Li2S-P2S5体系玻璃态电解质材料在室温下的离子电导率主要在10-4~10-3 S·cm-1范围[7, 8]。2004年,Murayama等[9]采用熔体淬火方法,以Li2S、P2S5为原料,在700℃的高温下锻烧8h,然后慢慢冷却至室温制备了组成范围0≤x≤0.27的Li3+5xP1-xS4新固溶体。结果发现,在x=0.065时,27℃下有最高的离子电导率1.5×10-4S·cm-1,活化能为22kJ·mol-1,而x=0.00时的电导率最低,说明在Li3PS4中通过Li+取代部分P5+而产生额外的锂离子导致离子电导率的大幅增加。
Minami等[10]研究了不同的熔化温度对70Li2S-30P2S5离子电导率的影响。结果表明,当熔化温度从750℃上升到900℃,会导致P2S74-转化成P2S64-,从而形成低离子电导率的结晶相Li4P2S6。因此,熔化温度在750℃时所获得的产物具有2.1×10-3S·cm-1的最高电导率,且锂离子迁移数趋近于1。Liu等[11]探究了以Ge0.35Ga0.05Q0.60(Q=S、Se)部分替换P2S5对0.5Li2S-xP2S5-(1-x)Ge0.35Ga0.05Q0.60(x=0.1,0.3,0.5,0.7,0.9)电解质的电化学稳定性的影响。结果表明,x≤0.5时,电导率随着Ge0.35Ga0.05Q0.60组分的增加而不断提高;进一步增加Ge0.35Ga0.05Q0.60的含量,电导率开始下降。在x=0.5时,掺杂Ge0.35Ga0.05Se0.60的样品(0.5Li2S·0.5P2S5·0.5Ge0.35Ga0.05Se0.60)有最高的离子电导率(2.9×10-4S·cm-1)。与之相比,掺杂Ge0.35Ga0.05S0.60的样品的电导率要低一个数量级。这是因为Se2-比S2-更有利于锂离子传导。表 1列出了采用熔体淬火法制备的硫化物电解质的离子电导率。
 下载:
导出CSV
下载:
导出CSV
| 原材料 | 组成 | 热处理条件 | 离子电导率/(S·cm-1) | 文献 |
| Li2S、P2S5 | Li3.325P0.935S4 | 700℃/8h | 1.5×10-4 | [9] |
| Li2S、P2S5 | 70Li2S-30P2S5 | 750℃/20h | 2.1×10-3 | [10] |
| Li2S、P2S5、Ge0.35Ga0.05Se0.60 | 0.5Li2S·0.5P2S5·0.5Ge0.35 Ga0.05Se0.60 | 750℃/10h | 1.5×10-3 | [11] |
| Li2S、P2S5、P2O5 | 70Li2S·27P2S5·3P2O5 | 750℃/10h | 3.0×10-3 | [13] |
| Li2S、P2S5、GeS2 | Li3.25Ge0.25P0.75S4 | 700℃/8h | 2.2×10-3 | [14] |
| Li2S、P2S5、LiCl、SnS | Li2S·P2S5·SnS·LiS | 600℃/8h | 5.89×10-4 | [12] |
| Li2S、P2S5、GeS2 | Li10GeP2S12 | 550℃/8h | 1.2×10-2 | [6] |
高能球磨法是制备硫化物电解质常用方法之一,原料在球磨过程中需经历混合、粉碎、非晶化和固态反应等过程,整个过程在室温下进行。通过该方法很难获得均匀的化学计量组成,因为原材料在研磨过程中容易残留在球和罐的表面,并且硫在退火过程中具有高挥发性,导致所得硫化物固体电解质在化学计量上存在差异[15]。相比于熔体淬火法,高能球磨法是一种固态扩散反应,其反应条件可控、安全性高、操作简单且球磨后获得的玻璃态样品只需在氩气气氛、较低温度下热处理即可获得玻璃-陶瓷态样品等优点。Hayashi等[16]分别利用ZrO2介质(500个球、直径为4mm)和Al2O3介质(10个球、直径为10mm)球磨混合Li2S、P2S5、P2S3原料制备了70Li2S-(30-x)-P2S5-xP2S3,该玻璃态样品经热处理得到了玻璃-陶瓷态样品。实验发现,使用ZrO2介质制备的玻璃-陶瓷态样品的离子电导率比以Al2O3介质制备的要高,同时发现,使用较小的氧化锆球可合成高电导率的玻璃-陶瓷态电解质。相较于纯玻璃态样品,玻璃-陶瓷态样品的导电性提高归功于高结晶度Li7P3S11的生成。
高能球磨法制备玻璃态硫化物电解质在不同温度下热处理所获得的样品,其结构和成分也各不相同。Tatsumisago等[17]以Li2S、P2S5为原料,采用高能球磨制备(100-x)Li2S-xP2S5玻璃态样品,然后经热处理获得了玻璃-陶瓷态样品。如图 1所示,70Li2S-30P2S5玻璃-陶瓷态有最高的电导率3.2×10-3 S·cm-1,这是由于热处理过程中形成了高电导率的新的亚稳态相Li7P3S11。80Li2S-20P2S5玻璃-陶瓷态电解质主要由类似于Li4-xGe1-xPxS4中高导电硫代-LISICON II相的晶体组成,因而在室温下可显示出高达10-3S·cm-1的电导率。以80Li2S-20P2S5为电解质,In或In-Li为负极,LiCoO2、Li4/3Ti5/3O4或非晶形V2O5为正极组装全固态锂离子电池,由图 1(b)第300圈的充放电曲线可得,使用不同电极的全固态电池有高的容量且100%的库伦效率,说明玻璃-陶瓷态电解质与各种固态电池正极材料相容,具有良好的循环性。
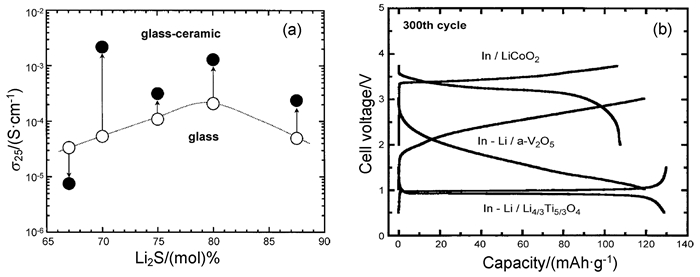
Mizuno等[18]采用高能球磨法制备xLi2S-(100-x)P2S5玻璃态样品,再将其在不同温度下热处理获得玻璃-陶瓷态样品。结果表明,热处理温度达到第一次结晶温度时,70Li2S-30P2S5玻璃-陶瓷态样品电导率为2.2×10-3S·cm-1,活化能为18kJ·mol-1。比较在240、360、550 ℃下热处理的结果,发现玻璃态样品在360℃下热处理获得的玻璃-陶瓷态样品有最高的电导率3.2×10-3S·cm-1,进一步提高热处理温度反而导致所得样品的电导率降低。
湿化学法是以液体溶剂作为介质的合成方法。相比于高能球磨法,该方法具有低能耗、省时和混合效果良好等优点,其所制备的颗粒粒径较小、孔隙少。Ito等[19]把Li2S、P2S5原料溶解在1, 2-二甲氧基乙烷溶剂(沸点为85℃)中后再放入蒸发器中去除有机溶剂,然后在真空干燥箱中180℃进行干燥,所得粉末分别在温度为200、250、300 ℃下加热1h得到70Li2S-30P2S5玻璃-陶瓷态样品。通过XRD发现,采用湿化学法可以成功合成具有Li7P3S11晶体相的硫化物电解质,遗憾的是在室温下最高电导率只有2.7×10-4S·cm-1,比高能球磨法合成的低。Xu等[20]分别使用了四氢呋喃、乙腈、四氢呋喃/乙腈混合溶液作为溶剂,去除溶剂后获得的粉末在250℃下烧结2h。通过乙腈溶剂合成的电解质在25℃下有最高电导率(9.7×10-4S·cm-1)、低活化能(31.2kJ·mol-1)以及宽而稳定的电化学窗口(5V)。目前已报道使用过的有机溶剂有N-甲基甲酰胺[21, 22]、乙腈[23, 24]、四氢呋喃[20]、乙酸乙酯[25]、1, 2-二甲氧基乙烷[19]和乙醇[26]等。采用湿化学法制备的硫化物电解质的电导率比球磨合成的要低,其原因可能是有机溶剂的残余和样品中有非晶相导致。但对于大规模生产而言,其耗能和反应时间减少。因此湿化学法在未来具有较大发展空间。
简而言之,熔融淬火法具有设备要求简单和投入低的特点,可用于制备玻璃类、陶瓷类和玻璃-陶瓷类材料,但还存在合成温度高,安全性低,持续时间长,产物组成较难控制的缺点。高能球磨法具有较高的安全系数和操作简单的优点,其不足在于耗时长且对原材料有损耗,一定程度会破坏材料的形貌。与前两种方法相比,湿化学法的合成更灵活,且在能耗和减少反应时间方面具有优势,在未来研究当中具有较大的提升空间。
从提高Li2S-P2S5体系电解质稳定性(化学稳定性和电化学稳定性)和离子电导率两方面考虑,可实施掺杂措施,主要掺杂物包括氧化物、硫化物和卤化物。引入的不同价态元素可形成不平衡的价态体系,从而产生新的空隙增加可迁移的离子浓度以提高离子电导率。掺杂还可通过改善主体结构的稳定性,弱化骨架与离子间作用力以利于离子的迁移,从而获得高离子电导率的导体材料。由于高能球磨法操作简单和反应条件可控,因此被广泛应用于掺杂改性。
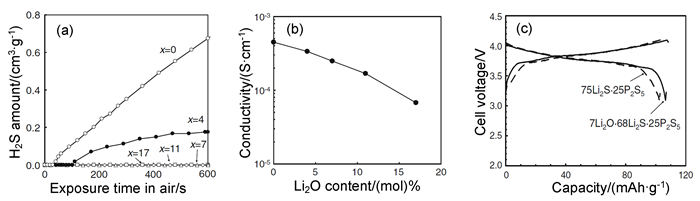
Li2S-P2S5体系的电解质的化学稳定性较差,容易吸收水分产生H2S气体。通过部分氧化物(Li2O或P2O5)替换硫化物来改变Li2S-P2S5的组成,可有效抑制H2S气体的产生;也可以通过掺杂其他金属氧化物作为H2S的吸收剂,增强其化学稳定性。Ohtomo等[27, 28]探究了采用一步球磨(Li2S、P2S5和Li2O一起球磨40h)和二步球磨(先将Li2S和P2S5球磨20h,再与Li2O放一起球磨20h)分别制备不同组分的xLi2O-(100-x)·(0.7Li2S·0.3P2S5)(x=17,20和25)玻璃态电解质,并用H2S传感器测量试样产生的H2S的量。结果发现,Li2O的添加可以有效减少H2S气体产生量,尤其是采用两步球磨制备的电解质,其在空气中水解产生的H2S气体产生量小于0.009cm3·g-1。他们进一步利用二步球磨法探究Li2O的添加量对75Li2S-25P2S5体系玻璃态电解质的在空气中产生H2S气体量的影响,分别制备了不同组分的xLi2O-(75-x)Li2S·25P2S5(x=0、4、7、11、17)电解质。如图 2所示,可发现x≥7的玻璃态电解质H2S气体产生量小于0.009cm3·g-1,远低于未掺杂样品的0.061cm3·g-1,说明替换Li2S可以有效抑制H2S气体的产生。遗憾的是,Li2O的加入会降低电解质的电导率,使用7Li2O·68Li2S·25P2S5组装的C/LiCoO2电池的放电容量更高。
此外,掺杂也可以提高Li2S-P2S5体系电解质的电化学稳定性。未掺杂Li2S-P2S5体系的电解质一般工作电压区间为5V,通过掺杂可有效扩展电化学窗口。Ujiie等[29]在70Li2S-30P2S5电解质中添加LiI,采用高能球磨制备(100-x)(0.7Li2S-0.3P2S5)·xLiI(0≤x≤20)电解质。结果发现,添加LiI后得到的玻璃态样品的电导率提高,而玻璃-陶瓷态样品的电导率却下降了,可能是因为产生的未知晶体具有低电导率特性所致。由80(0.7Li2S-0.3P2S5)·20LiI玻璃态电解质的循环伏安曲线可知,在电位范围1.0~10 V之间没有明显阴极或阳极峰,表明固态电解质在高电位下具有良好的稳定性。Minami等[13]在70Li2S-30P2S5电解质中添加P2O5,通过循环伏安曲线,可以得知添加了P2O5,在整个电位-0.1~10 V范围内,除了锂沉积和溶解对应的电流峰,并没有其他大的电流峰,而未掺杂70Li2S-30P2S5玻璃-陶瓷态电解质却有一个额外的表示游离S2-的氧化的阳极电流峰出现。可见,用P2O5替换部分P2S5后的产物显示出更高的电化学稳定性。表 2总结了不同组分硫化物电解质的电化学窗口,从中看出掺杂可有效扩展硫化物固态电解质的电化学窗口。
下载:
导出CSV
| 组成 | 电化学窗口/V | 种类 | 电导率/(S·cm-1) | 文献 |
| 70Li2S-30P2S5 | 5 | glass | 1.58×10-3 | [35] |
| 80Li2S-20P2S5 | 5 | glass-ceramic | 4.3×10-4 | [32] |
| 90Li3PS4·10ZnO | 5 | glass | ﹥10-4 | [31] |
| Li7P2.9S10.85Mo0.01 | 5 | glass-ceramic | 4.8×10-3 | [33] |
| 2.25Li3+0.55P1-0.11S4·0.2LaS2 | 8 | glass-ceramic | 7.41×10-5 | [37] |
| 70Li2S·27P2S5·3P2O5 | 10 | glass-ceramic | 3.0×10-3 | [13] |
| 75Li2S-24P2S5-P2O5 | 10 | glass-ceramic | 8.0×10-4 | [34] |
| Li10GeP2S11.7O0.3 | 10 | crystalline | 1.03×10-2 | [36] |
| 80(0.7Li2S-0.3P2S5)·20LiI | 10 | glass | 5.6×10-4 | [29] |
| 95(0.8Li2S-0.2P2S5)·5LiI | 10 | glass-ceramic | 2.7×10-3 | [30] |
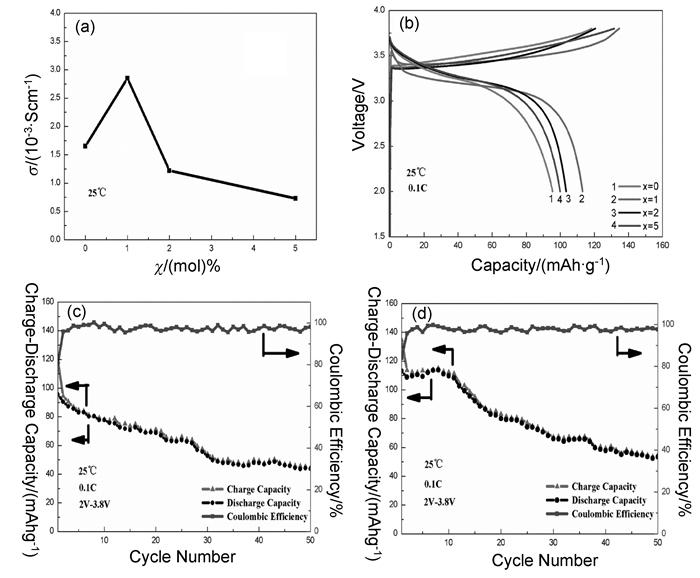
对全固态锂离子电池来说,高离子电导率是决定电解质实用性的主要条件之一。Lu等[38]采用高能球磨制备了不同组分的玻璃态和玻璃-陶瓷态的(100-x)(70Li2S-30P2S5)-xLi2ZrO3(x=0、1、2、5)电解质,研究了不同含量的Li2ZrO3掺杂、热处理温度以及时间对固态电解质的电化学性能的影响。如图 3(a)所示,随着Li2ZrO3含量的增加,玻璃态电解质的电导率不断增大,当x=1时,99(70Li2S-30P2S5)-Li2ZrO3电导率最大2.85×10-3S·cm-1。不同的热处理温度对99(70Li2S-30P2S5)-Li2ZrO3玻璃态电解质也有很大的影响,结果发现,热处理温度低于275℃的样品电导率低于1.0×10-3S·cm-1;随着热处理温度升高至285℃,样品的电导率升至2.85×10-3S·cm-1;进一步升高温度到295℃时,样品的电导率重新降低至1.04×10-3S·cm-1。另一方面,在热处理温度为285℃,不同的保温时间下99(70Li2S-30.P2S5)-Li2ZrO3电解质所获得的电导率先随保温时间增加而增加,然后又不断减少,优化的保温时间为4h。从图 3(b,c,d)可知,采用99(70Li2S-30P2S5)-Li2ZrO3作为LiCoO2/SE/In-Li的电解质,相比使用70Li2S-30P2S5作为电解质,其放电容量在第10次和第50次的充放电循环分别提高了41.0%和21.9%。交流阻抗(EIS)结果表明,掺杂Li2ZrO3可以减少固态电解质和电极之间的界面电阻,这归因于Li2ZrO3本身具有稳定的晶体结构和高锂离子扩散系数。
Xu等[33]以Li2S、P2S5、MoS2为原料通过高能球磨后热处理制备了Li7P2.9S10.85Mo0.01和Li7P3S11玻璃-陶瓷态电解质,并组装了Li-S电池进行测试。结果表明,在Li2S-P2S5玻璃-陶瓷态电解质中掺杂MoS2可以提高电导率,其在室温下电导率达到4.8×10-3 S·cm-1,电化学性能稳定,其作为电解质的Li-S电池展现了比较高的放电容量(1020mAh·g-1)和更好的循环性能;而以Li7P3S11作为电解质的Li-S电池放电容量只有775mAh·g-1,多次循环后容量快速下降。原因在于掺杂MoS2可以提供大量缺陷并拓宽锂离子迁移通道,有利于锂离子快速迁移。表 3总结了已报道的各种多元硫化物电解质在室温下的离子电导率,从中可以发现经不同物质掺杂改性后的电解质离子电导率相较于未掺杂样品均有较大提高。
下载:
导出CSV
| 组成 | 电导率/(S·cm-1)a | 种类 | 文献 |
| 99(70Li2S·30P2S5)·Li2ZrO3 | 2.85×10-3(1.65×10-3) | glass-ceramic | [38] |
| Li7P2.9S10.85Mo0.01 | 4.8×10-3(2.6×10-3) | glass-ceramic | [33] |
| Li3.05Ge0.05P0.95S4 | 1.2×10-3(9.4×10-4) | glass-ceramic | [39] |
| 98(70Li2S·30P2S5)·2GeS2 | 5.4±0.1×10-3(4.2×10-3) | glass-ceramic | [45] |
| 95(0.8Li2S·0.2P2S5)·5LiI | 2.7×10-3(8.2×10-4) | glass-ceramic | [30] |
| 80(0.7Li2S·0.3P2S5)·20LiI | 5.6×10-4(1.3×10-4) | glass | [29] |
| 90(0.7Li2S·0.3P2S5)·10LiBr | 6.5×10-3(5.2×10-3) | glass-ceramic | [40] |
| 70Li2S·29P2S5·P2S3 | 5.4×10-3(4.2×10-3) | glass-ceramic | [41] |
| 70Li2S·27P2S5·3P2O5 | 4.6×10-3(4.2×10-3) | glass-ceramic | [41] |
| 97(0.78Li2S·0.22P2S5)·3Li3BO3 | 1.03×10-3(6.14×10-4) | glass-ceramic | [42] |
| 30LiI·70(0.07Li2O·0.68Li2S·0.25P2S5) | 1.3×10-3(~5×10-4) | glass | [43] |
| 86.9Li3PS4·13.1LiAlS2 | 6.0×10-4(2×10-4~3×10-4) | glass | [44] |
| 注:a括号里的数值代表未掺杂样品的离子电导率值。 | |||
从锂离子电池组成来看,除固态电解质本身性能以外,电池的最终性能还和所组成电极材料有关。目前主要面临如何构筑具有高效的离子-电子通道的复合正极和电极/硫化物电解质的界面问题。
全固态电池中关键的组成之一是正极,不同于传统正极片,其主要由正极活性物质、固态电解质以及导电剂组成,其中添加固态电解质和导电剂是为了加强正极中的锂离子扩散和电子电导率。Mizuno等[46]分别探究了以乙炔黑(AB)、气相生长碳纤维(VGCF)、TiN、Ni作为导电添加剂与LiCoO2活性物质组成的复合正极片对以Li2S-P2S5玻璃-陶瓷态为电解质和In为负极的全固态电池的性能的影响。结果发现,相比于TiN、Ni,碳材料能更加有效减少复合正极片的电阻。在电流密度为64μA·cm-2下,以VGCF为导电剂的电池在充放电循环超过50圈后,其放电容量仍然可以维持在大约100mAh·g-1,高于以AB、TiN、Ni为导电添加剂的电池性能。在电流密度超过1mA·g-1情况下仍得到了相似的结果,这是由于VGCF由亚微米级有序纤维组成,可作为桥梁使电极和电解质之间实现紧密的固-固接触,并且VGCF在复合正极中形成比AB更连续的电子传导路径。表 4总结了已报道的由含不同导电剂正极与固态电解质组成的固态电池的性能。除正极极片本身组成外,其制备工艺也可影响相关固态电池的性能。现阶段,通常使用干混方法制备正极极片,在此过程中固体颗粒很难均匀混合与分散,如AB作为导电剂时在干混过程中很容易团聚[47]。采用湿法混合可以得到各组分更均匀和结果重现性好的正极片。Kim等[48]对比制备全固态电池的复合正极采用干混与湿混的差别,结果发现,虽然采用干混制备复合正极较湿法混合得到的极片具有更高的首次充放电容量(127mAh·g-1),但是其循环20圈后的容量减到54mAh·g-1,远低于湿法混合电极的81mAh·g-1(初始容量为111mAh·g-1),循环性能的提高是因为湿法混合使组成颗粒更均匀分布,改进了颗粒间的接触。
下载:
导出CSV
| 固态电解质 | 电导率/(S·cm-1) | 活性物质 | 导电剂 | 固态电解质/活性物质/导电剂(质量比) | 放电容量/(mAh·g-1) | 文献 |
| 70Li2S-30P2S5 | 1.58×10-3 | Li2S | Super P+VGCF | 30/60/6.7+3.3 | 1055(1st) | [35] |
| Li7P2.9S10.85Mo0.01 | 4.8×10-3 | S | Super P | 60/30/10 | 1020(1st) | [33] |
| 99(70Li2S·30P2S5)·Li2ZrO3 | 2.85×10-3 | LiCoO2 | 3/8 | 113(1st) | [35] | |
| 30LiI·70(0.07Li2O·0.68Li2S·0.25P2S5) | 1.3×10-3 | LiCoO2/LiNbO3 | 30/70 | 116(30st) | [40] | |
| 97(0.78Li2S·0.22P2S5)·3Li3BO3 | 1.03×10-3 | LiCoO2 | Super P | 19.6/78.4/2 | 149(40st) | [39] |
| Li3.05Ge0.05P0.95S3.9Se0.1 | 1.4×10-3 | LiCoO2 | Super P | 30/20/3 | 62(10st) | [50] |
| 70Li2S·29P2S5·P2S3 | 5.4×10-3 | Li4Ti5O12 | VGCF | 60/40/4 | 140(700st) | [38] |
| Li10GeP2S12 | 1.2×10-2 | LiNi0.5Mn1.5O4/LiNbO3 | Super P | 57.5/38.5/4 | 80(1st) | [51] |
| 78Li2S-22P2S5 | 2.0×10-4 | LiCoO2/Li2CO3 | Super-P | 34/64/2 | 137.4(1st) | [52] |
| 80Li2S-20P2S5 | FePS3 | 107(30st) | [49] | |||
| 78Li2S-22P2S5 | 8.5×10-4 | LiCoO2 | Super P | 59/39/2 | 81(20st) | [48] |
不同于大多数报道,Fujii等[49]发现,FePS3的电子电导率大约为10-5S·cm-1,而且是层状结构,有利于锂离子迁移。因此在全固态锂离子电池中直接使用FePS3作为正极复合材料且复合正极材料中没有添加固态电解质和导电添加剂。电池在30圈充放电循环后,放电容量为107mAh·g-1,说明使用没有添加固态电解质和导电剂的复合正极材料的电池也可以运行。这是因为FePS3含有大量的供锂离子迁移和电子传导的通道。综合来讲,运用具有高效的离子/电子通道的活性正极材料,改善活性物质与固态电解质、导电剂的分散状态对于有效利用活性物质和提升固态电池的性能尤其重要。
电极与电解质的界面问题是全固态电池发展的关键问题之一,主要分为物理和化学两方面。物理方面主要为接触问题。电解质与电极之间维持面接触容易产生气孔和裂缝等缺陷,从而限制了锂离子在界面处的传输;并且,锂离子在传输过程中界面处的体积膨胀对固-固界面的稳定性提出更高的要求。化学方面,电解质与电极发生副反应,使得固-固界面稳定性降低,界面阻抗增大,无法实现锂离子的快速迁移[53]。如正极活性物质与电解质之间形成空间电荷层、电极与电解质的副反应、电解质与电极之间元素的相互扩散等。
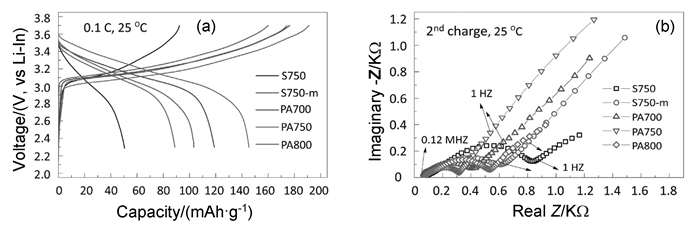
Peng等[54]探究了LiNi0.8Co0.15Al0.05O2正极材料的颗粒尺寸、表面残余和缺陷跟电化学性能的关系。利用球磨技术虽然可以除去LiNi0.8Co0.15Al0.05O2表面的残余碳酸盐并降低颗粒尺寸,但同时带来了表面缺陷,其表面缺陷可以通过退火过程恢复。以Li10GeP2S12作为电解质组装NCA/Li-In全固态电池,测试结果如图 4,发现使用先球磨后退火(在750℃下5h)合成的NCA正极材料(标记为PA750)组装的全固态电池,首次放电容量由51mAh·g-1提高到146mAh·g-1,同时库伦效率也得到极大的提高,由交流阻抗谱发现先球磨后退火可以极大降低界面阻抗。
Oh等[51]发现,以LiNi0.5Mn1.5O4为正极、Li10GeP2S12电解质和Li负极所组成的固态电池的首次放电容量只有80mAh·g-1,且在不断充放电循环中放电容量逐渐减少,由X射线衍射和电化学阻抗谱测量得知,这是由于正极复合材料上的电阻界面层的生长引起的。通过在LiNi0.5Mn1.5O4正极表面涂覆一层LiNbO3缓冲层,有效抑制了界面阻抗层的形成。目前使用的缓冲层材料有Li2O-SiO2[55]、LiNbO3[51]、Li4SiO4-Li3PO4[56]、CoS、NiS[57]等。
大多数正极材料是氧化物,由于硫化物电解质与正极材料之间的化学势不同,导致电解质中锂向电极氧化物中移动,造成界面处硫化物固态电解质“贫Li”,载流子数目减少,产生较大的阻抗,而且当氧化物电极只是离子导体时,形成的空间电荷层不仅存在于电解质中,也存在于氧化物中[58]。为了抑制空间电荷层的形成,可以在正极表面涂覆一层保护层。Ohta等[59]在正极LiCoO2颗粒上喷涂不同厚度的Li4Ti5O12,通过观察首次充电曲线的斜率变化证实Li4Ti5O12缓冲层对空间电荷层的形成产生抑制作用。随着厚度的增加,首次充电曲线的效率不断增加,说明空间电荷层逐渐消失,但厚度增加到一定程度时,虽然空间电荷层消失了,可界面阻抗逐渐增大,这是由于Li4Ti5O12的低离子电导率所造成。
金属锂是可充电电池的理想负极,它具有质轻、氧化还原电位低(Li/Li+相对于标准氢电极标准氧化还原电位为-3.04V)和极高的理论比容量(3860mAh·g-1)等特点。当作为全固态锂离子电池负极时,其很强的还原性容易使固体电解质中某些高价态金属阳离子得电子而被还原,并生成一层高电阻界面相。目前,主要通过电解质表面修饰、形成锂和其他元素的合金以及增加聚合物隔膜三种途径来解决问题。Sakuma等[60]探究了以Li4-xGe1-xPxS4为电解质以及Li-M (M=Sn,Si)合金为负极的全固态电池的电化学性能,并与由Li所组成的电池性能进行了对比。结果表明,Li-M(M=Sn,Si)合金具有比Li更高的氧化还原电位,可减少副反应发生,有效抑制界面电阻的增长。然而,以Li为负极的电池中,随着电解质材料中Ge含量的升高,副反应越严重,界面阻抗不断增大。Ogawa等[61]用激光脉冲沉积法在金属锂的表面沉积了一层厚度为20nm的Si,和LiCoO2正极以及Li2S-P2S5电解质组成电池进行了电化学性能测试。结果显示,Si层有效抑制了Li对固态电解质的还原,从而显著提高了电池的循环性能。沉积Si层后的电池在循环1000圈后,容量保持率接近100%,远高于未沉积Si层的全固态电池的76%。
表征Li2S-P2S5体系电解质传输性质的主要参数为电导率和离子迁移数,常用交流阻抗法和稳态电流法分别进行测量。
电导率是表征硫化物电解质离子导电能力的重要指标,反映了体系中全部离子的整体迁移率,决定着锂离子电池所能提供的最大电能。通常硫化物电解质的电导率由Arrhenius方程来表述:
|
$ \sigma=A T^{-1} \exp \left(-E_{\mathrm{a}} / R T\right) $ |
式中,σ为硫化物电解质的电导率,Ea为电导活化能,A为指前因子,T为硫化物电解质电导率测定时的温度。可以看出,电解质的电导率与其工作温度有关,温度越高,则导电性能越好。
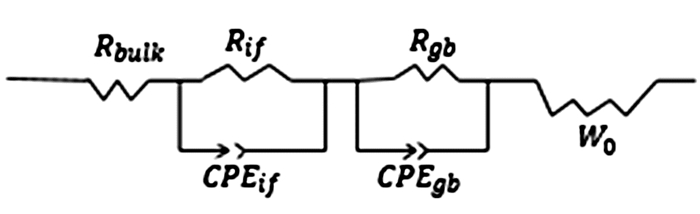
EIS法主要用于研究电极过程动力学和表面现象,可用来测定固态电解质的离子电导率。通常,电解质被夹在两片电极中间进行测试。电极材料中的原子通过氧化还原变成与电解质中导电离子相同的离子,为非阻塞电极,如金属锂[36, 50];另一类电解质中导电离子不能穿过电极/电解质界面,为阻塞电极,如铟箔[19, 20, 33]、不锈钢[5, 62]、碳浆[10, 34]、金膏[44]。两类电极的双电层建立方式不同,因此等效电路也不同。使用阻塞电极时,电解质中的导电离子(Li+)无法穿越电极/电解质界面,只能在电场力作用下向一个界面富集,形成化学梯度,并导致带负电荷的离子向相反方向迁移。图 5是它的等效电路,其中CPE可认为是一个漏电容,它的性质介于电阻与电容之间,是由于电极/电解质接触界面的不平整性所造成。图中的CPEif表示不平整接触界面的电容效应和界面离子扩散阻抗的总和,CPEgb表示硫化物电解质的法拉第容量和电解质内离子传递阻抗的总和,Rbulk、Rif、Rgb分别为电解质的本体电阻、电解质与电极的界面电阻和晶界电阻,Wo为Warburg扩散阻抗。在非阻塞型电极组成的测试电池中,离子可以穿过电极/电解质界面,于是在此界面形成了电荷传递电阻Rct。因Rct与双电层电容并联,等效电路相对复杂,故测试电导率一般选用阻塞型电极。
经EIS测试,硫化物电解质的电导率可由以下方程计算得出:
|
$ \sigma=\frac{d}{R_{\text {total }} S} $ |
式中,σ为硫化物电解质的电导率,d为硫化物电解质压片的厚度,Rtotal为总的阻抗(电解质本体阻抗、晶界阻抗和界面阻抗之和),S为电极与硫化物电解质的接触面积。
离子迁移数是指某一离子所运载的电流占总电流的比值,反映了与某种元素有关的离子迁移率,决定着锂离子电池所能提供的最大电流密度和可充电电池的使用寿命,是设计电池时选择电解质的重要参数之一。在电场作用下,电解质体系中正负两类离子可以同时运动,这样将形成电解质盐的浓度梯度,产生与所加电场反向的浓差极化电势,导致系统的电化学性能下降。因此,若锂离子的迁移数趋于1且阴离子的迁移数很小,说明硫化物电解质中的离子传导主要由Li+来完成,扩散导电的作用就很小,可有效消除电池内电解质的浓差极化,为电池提供最大的极限电流。测定电解质锂离子迁移数的方法包括离子迁移数法[63]、稳态电流法[64]、Tubandt重量分析法[65]和交流阻抗方法[66]等,最常用的为稳态电流法。该方法是一种电势阶跃法和交流阻抗法联用的方法:将硫化物电解质组装成非阻塞Li/电解质/Li模拟电池,利用电势阶跃法对所构成的电池施加一小幅度恒定直流极化电压ΔV,同时记录电流随时间的变化;在开始极化的瞬间,起始电流(I0)最大,体系中所有可迁移的组分(包括正负离子和离子聚集体等)对电荷的传输过程都有贡献。随着极化的进行两电极之间的浓度梯度逐渐形成,浓度梯度的产生,降低了阴、阳离子的迁移率,电流呈迅速减小的趋势,最终形成了一个稳定的浓度梯度。此时,阴离子的迁移被抑制,仅有锂离子的迁移和漏电流(通过电子和空穴形成),所记录电流趋于某一定值,称之为稳态电流(Is)。稳态电流反映了锂离子定向迁移对电荷传输的贡献。
另一方面,可使用不锈钢作为阻塞电极,同样施加一小幅度恒定直流极化电压ΔV,由于锂离子不能穿过电极/电解质界面,因此起始电流(I0)比较小,经过极化后漏电流趋于稳定。根据漏电流的大小可以判断电子和空穴对电导的贡献。综合来讲,稳态电流法具有测定方便、原理简明、所得结果重现性好的优点,因此在研究硫化物电解质中被广泛应用。
固态锂离子电池是未来锂离子电池的重要发展方向之一,提高固态电解质的电导率是需要解决的主要核心问题,其关键在于材料的合成。其中,湿化学法具有很大发展空间,将成为未来合成固态电解质的主要研究方法,其关键在于所得产物组成的控制和溶剂的去除。鉴于固态电解质在提高离子电导率与加强稳定性过程存在矛盾,因此选择一种能提高离子电导率并同时加强其稳定性的掺杂剂显得尤为重要。从锂离子固态电池整个体系来看,部分电解质的电导率已经接近液态电解质所能达到的程度,但与电极之间的界面问题极大地限制了其应用。因此,围绕特定固态电解质发展合适的正极和负极体系将对电池性能和应用有重要影响。尽管全固态锂离子电池有诸多问题,但因其具有传统锂离子电池无法比拟的优点,预期其未来将会得到广泛的应用。
N Armand, J M Tarascon. Nature, 2008, 451:652~657. doi: 10.1038/451652a
P Simon, Y Gogotsi. Nat. Mater., 2008, 7:845~854. doi: 10.1038/nmat2297
M Y Gao, C C Shih, S Y Pan et al. J. Mater. Chem. A, 2018, 6(42):20546~20563. doi: 10.1039/C8TA07246A
M Tatsumisago, A Hayashi. Int. J. Appl. Glass Sci., 2014, 5(3):226~235. doi: 10.1111/ijag.12084
J Kim, Y Yoon, J Lee et al. J. Power Sources, 2011, 196:6920~6923. doi: 10.1016/j.jpowsour.2010.12.020
N Kamaya, K Homma, Y Yamakawa et al. Nat. Mater., 2011, 10:682~686. doi: 10.1038/nmat3066
M Tatsumisago, K Hirai, T Minami et al. J. Ceram. Soc. Jpn., 1993, 101(11):1315.
S Kondo, K Takada, Y Yamamura. Solid State Ionics, 1992, 53~56:1183~1186. doi: 10.1016/0167-2738(92)90310-L
M Murayama, N Sonoyama, A Yamada et al. Solid State Ionics, 2004, 170:173~180. doi: 10.1016/j.ssi.2004.02.025
K Minami, F Mizuno, A Hayashi et al. Solid State Ionics, 2007, 178:837~841. doi: 10.1016/j.ssi.2007.03.001
Z Liu, Y Tang, Y Wang et al. J. Power Sources, 2014, 260:264~267. doi: 10.1016/j.jpowsour.2014.03.036
X Sun, Y Sun, F Cao et al. J. Alloys Compd., 2017, 727:1136~1141. doi: 10.1016/j.jallcom.2017.08.167
K Minami, A Hayashi, M Tatsumisago. Solid State Ionics, 2008, 179:1282~1285. doi: 10.1016/j.ssi.2008.02.014
R Kanno, M Murayama. J. Electrochem. Soc., 2001, 148:A742~A746. doi: 10.1149/1.1379028
S Chen, D Xie, G Liu et al. Energy Storage Mater., 2018, 14:58~74. doi: 10.1016/j.ensm.2018.02.020
A Hayashi, K Minami, S Ujiie et al. J. Non-Crystal Solids, 2010, 356:2670~2673. doi: 10.1016/j.jnoncrysol.2010.04.048
M Tasumisago, F Mizuno, A Hayashi. J. Power Sources, 2006, 159:193~199. doi: 10.1016/j.jpowsour.2006.04.037
F Mizuno, A Hayashi, K Tadanaga et al. Solid State Ionics, 2006, 177:2721~2725. doi: 10.1016/j.ssi.2006.04.017
S Ito, M Nakakita, Y Aihara et al. J. Power Sources, 2014, 271:342~345. doi: 10.1016/j.jpowsour.2014.08.024
R C Xu, X H Xia, Z J Yao et al. Electrochim. Acta, 2016, 219:235~240. doi: 10.1016/j.electacta.2016.09.155
S Teragawa, K Aso, K Tadanaga et al. J. Mater. Chem. A, 2014, 2:5095~5099. doi: 10.1039/c3ta15090a
S Teragawa, K Aso, K Tadanaga et al. J. Power Sources, 2014, 248:939~942. doi: 10.1016/j.jpowsour.2013.09.117
X Y Yao, D Liu, C S Wang et al. Nano Lett., 2016, 16:7148~7154. doi: 10.1021/acs.nanolett.6b03448
M Calpa, N C R Navarro, A Miura et al. RSC Adv., 2017, 7:46499~46504. doi: 10.1039/C7RA09081A
N H H Phuc, M Totani, K Morikawa et al. Solid State Ionics, 2016, 288:240~243. doi: 10.1016/j.ssi.2015.11.032
S Yubuchi, S Teragawa, K Aso et al. J. Power Sources, 2015, 293:941~945. doi: 10.1016/j.jpowsour.2015.05.093
T Ohtomo, A Hayashi, M Tatsumisago et al. J. Non-Cryst. Solids, 2013, 364:57~61. doi: 10.1016/j.jnoncrysol.2012.12.044
T Ohtomo, A Hayashi, M Tatsumisago et al. J. Solid State Electrochem., 2013, 17:2551~2557. doi: 10.1007/s10008-013-2149-5
S Ujiie, A Hayashi, M Tatsumisago. Solid State Ionics, 2012, 211:42~45. doi: 10.1016/j.ssi.2012.01.017
S Ujiie, A Hayashi, M Tatsumisago. J. Solid State Electrochem., 2013, 17:675~680. doi: 10.1007/s10008-012-1900-7
A Hayashi, H Muramatsu, T Ohtomo. J. Mater. Chem. A, 2013, 1:6320~6326. doi: 10.1039/c3ta10247e
F Mizuno, T Ohtomo, A Hayashi et al. Solid State Ionics, 2006, 177:2753~2757. doi: 10.1016/j.ssi.2006.04.024
R Xu, X Xia, X Wang et al. J. Mater. Chem., 2017, A5:2829~2834. https://pubs.rsc.org/en/content/articlelanding/2017/ta/c6ta10142a/unauth#!divAbstract
Y C Tao, S Chen, D Liu et al. J. Electrochem. Soc., 2016, 163(2):A96~A101. doi: 10.1149/2.0311602jes
Y Zhang, K Chen, Y Shen et al. Solid State Ionics, 2017, 305:1~6. doi: 10.1016/j.ssi.2017.03.024
Y Sun, K Suzuki, K Hara et al. J. Power Sources, 2016, 324:798~803. doi: 10.1016/j.jpowsour.2016.05.100
Z Q Liu, Y F Tang, X J Lyu et al. Ceram. Int., 2014, 40:15497~15501. doi: 10.1016/j.ceramint.2014.07.011
P Lu, F Ding, Z Xu et al. J. Power Sources, 2017, 356:163~171. doi: 10.1016/j.jpowsour.2017.04.083
J E Trevey, Y S Jung, S Lee. Electrochim. Acta, 2011, 56:4243~4247. doi: 10.1016/j.electacta.2011.01.086
S Ujiie, T Inagaki, A Hayashi et al. Solid State Ionics, 2014, 263:57~61. doi: 10.1016/j.ssi.2014.05.002
K Minami, A Hayashi, S Ujiie et al. Solid State Ionics, 2011, 192:122~125. doi: 10.1016/j.ssi.2010.06.018
M Eom, S Choi, S Son et al. J. Power Sources, 2016, 331:26~31. doi: 10.1016/j.jpowsour.2016.09.032
T Ohtomo, A Hayashi, M Tatsumisago et al. Electrochemistry, 2013, 81(6):428~431. doi: 10.5796/electrochemistry.81.428
Y Ooura, N Machida, M Naito et al. Solid State Ionics, 2012, 225:350~353. doi: 10.1016/j.ssi.2012.03.003
K Minami, A Hayashi, M Tatsumisago. J. Non-Cryst. Solids, 2010, 356:2666~2669. doi: 10.1016/j.jnoncrysol.2010.05.016
F Mizuno, A Hayashi, K Tadanaga et al. J. Electrochem. Soc., 2005, 152(8):A1499~A1503. doi: 10.1149/1.1939633
J K Hong, J H Lee, S M Oh. J. Power Sources, 2002, 111:90~96. doi: 10.1016/S0378-7753(02)00264-1
J Kim, M Eom, S Noh et al. Electron. Mater. Lett., 2012, 8(2):209~213. doi: 10.1007/s13391-012-2037-7
Y Fujii, A Miura, N C R Navarro et al. Electrochim. Acta, 2017, 241:370~374. doi: 10.1016/j.electacta.2017.04.089
J E Trevey, Y S Jung, S H Lee. J. Power Sources, 2010, 195:4984~4989. doi: 10.1016/j.jpowsour.2010.02.042
G Oh, M Hirayama, O Kwon et al. Chem. Mater., 2016, 28:2634~2640. doi: 10.1021/acs.chemmater.5b04940
J Kim, M Kim, S Noth et al. Ceram. Int., 2016, 42:2140~2146. doi: 10.1016/j.ceramint.2015.09.126
陈龙, 池上森, 董源等.硅酸盐学报, 2018, 46(1):21~34. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=gsyxb201801003
G Peng, X Yao, H Wan et al. J. Power Sources, 2016, 307:724~730. doi: 10.1016/j.jpowsour.2016.01.039
A Sakuda, H Kitaura, A Hayashi et al. Electrochem. Solid-State Lett., 2008, 11(1):A1~A3. doi: 10.1149/1.2795837
Y Sakurai, A Sakuda, A Hayashi et al. Solid State Ionics, 2011, 182:59~63. doi: 10.1016/j.ssi.2010.12.001
A Sakuda, N Nakamoto, H Kitaura et al. J. Mater. Chem., 2012, 22:15247~15254. doi: 10.1039/c2jm32490c
K Takada. IEICE Technical Report, 2008, 107:43~47.
N Ohta, K Takada, K Zhang et al. Adv. Mater., 2006, 18(17):2226~2229. doi: 10.1002/adma.200502604
M Sakuma, K Suzuki, M Hirayama et al. Solid State Ionics, 2016, 285:101~105. doi: 10.1016/j.ssi.2015.07.010
M Ogawa, R Kanda, K Yoshida et al. J. Power Sources, 2012, 205:487~490. doi: 10.1016/j.jpowsour.2012.01.086
Y Seino, T Ota, K Takada et al. Energy Environ. Sci., 2014, 7(2):627~631. doi: 10.1039/C3EE41655K
张虎成, 轩小朋, 王键等.电源技术, 2003, 27(1):54~57. doi: 10.3969/j.issn.1002-087X.2003.01.017
P G Bruce, C A Vincent. J. Electroanal. Chem., 1987, 225:1~17. doi: 10.1016/0022-0728(87)80001-3
I I Olsen, R Koksbang, E Skou. Electrochim. Acta, 1995, 40:1701~1706. doi: 10.1016/0013-4686(95)00094-U
P R Sorensen, T Jacobesn. Electrochim. Acta, 1982, 27:1671~1675. doi: 10.1016/0013-4686(82)80162-X

图 1 (a) 不同Li2S含量的玻璃态和玻璃-陶瓷态在室温下的电导率;(b) In或Li-In合金/80Li2S·20P2S5玻璃-陶瓷/LiCoO2、Li4/3Ti5/3O4或非晶形V2O5中所有固态电池在第300圈循环充放电曲线[17]
Figure 1 (a) Conductivity of glassy and glass-ceramic states with different Li2S contents at room temperature; (b) Charge-discharge curves at the 300th cycle for all-solid-state cells or In-Li alloy/80Li2S·20P2S5 glass-ceramic /LiCoO2, Li4/3Ti5/3O4 or amorphous V2O5[17]

图 2 (a) 玻璃粉末产的H2S气体量;(b)机械研磨制备的玻璃粉末的电导率;(c)75Li2S·25P2S5和7Li2O·68Li2S·25P2S5玻璃组装固态C/LiCoO2电池的充放电曲线(在室温、3.0~4.1 V的电压范围内、在0.1C的电流密度下进行测量)[28]
Figure 2 (a) Amounts of H2S gas generated from the glass powders; (b) Electrical conductivities of the glass powders prepared using mechanical milling at room temperature; (c) Charge and discharge curves of 75Li2S·25P2S5 and 7Li2O·68Li2S·25P2S5 glass assembled solid state C/LiCoO2 batteries (measured at room temperature, 3.0~4.1 V, at 0.1 C current density)[28]

图 3 (a) (100-x)(70Li2S-30P2S5)-xLi2ZrO3(x=0、1、2、5)玻璃态电解质在室温下的电导率;(b)全固态锂离子电池LiCoO2/(100-x)(70Li2S-30P2S5)-xLi2ZrO3/In-Li (x=0, 1, 2, 5)的首次充放电曲线;(c)x=0,(d)x=1全固态锂离子电池LiCoO2/(100-x)(70Li2S-30P2S5)-xLi2ZrO3/In-Li的循环性能[38]。
Figure 3 (a) The lithium-ion conductivities of (100-x)(70Li2S-30P2S5)-xLi2ZrO3 (x=0, 1, 2, 5) glass samples at room temperature; (b) The initial charge-discharge curves of all-solid-state lithium-ion batteries of LiCoO2/(100-x)(70Li2S-30P2S5)-xLi2ZrO3/In-Li (x=0, 1, 2, 5); The long cyclic performances of all-solid-state lithium-ion batteries of LiCoO2/(100-x)(70Li2S-30P2S5)-xLi2ZrO3/In-Li for 50 cycles. (c) x=0; (d) x=1[38]

图 4 (a) 以LiNi0.8Co0.15Al0.05O2粉末(S750、S750-m、PA700、PA750、PA800)组装全固态NCA/LGPS/Li-In电池的首次充放电曲线;(b)以LiNi0.8Co0.15Al0.05O2粉末(S750、S750-m、PA700、PA750、PA800)正极、Li-In负极的全固态电池(第二次充电电压达到3.7V)的交流阻抗谱[54]
Figure 4 (a) Initial charge/discharge curve for NCA powders S750, S750-m, PA700, PA750 and PA800; (b) Nyquist plots of S750, S750-m, PA700, PA750 and PA800 at the second charge state of 3.7 V vs Li-In alloy anode[54]

图 5 阻塞型电极组成的测试电池模拟等效电路
Figure 5 Equivalent circuit of a test cell with blocking electrodes
表 1 各种多元硫化物电解质的离子电导率(S·cm-1)
Table 1. Ionic conductivity of various polysulfide solid electrolytes (S·cm-1)
| 原材料 | 组成 | 热处理条件 | 离子电导率/(S·cm-1) | 文献 |
| Li2S、P2S5 | Li3.325P0.935S4 | 700℃/8h | 1.5×10-4 | [9] |
| Li2S、P2S5 | 70Li2S-30P2S5 | 750℃/20h | 2.1×10-3 | [10] |
| Li2S、P2S5、Ge0.35Ga0.05Se0.60 | 0.5Li2S·0.5P2S5·0.5Ge0.35 Ga0.05Se0.60 | 750℃/10h | 1.5×10-3 | [11] |
| Li2S、P2S5、P2O5 | 70Li2S·27P2S5·3P2O5 | 750℃/10h | 3.0×10-3 | [13] |
| Li2S、P2S5、GeS2 | Li3.25Ge0.25P0.75S4 | 700℃/8h | 2.2×10-3 | [14] |
| Li2S、P2S5、LiCl、SnS | Li2S·P2S5·SnS·LiS | 600℃/8h | 5.89×10-4 | [12] |
| Li2S、P2S5、GeS2 | Li10GeP2S12 | 550℃/8h | 1.2×10-2 | [6] |
 下载: 导出CSV
下载: 导出CSV
表 2 各种硫化物固态电解质的电化学窗口
Table 2. summary of the electrochemical windows of various sulfide-based solid electrolytes
| 组成 | 电化学窗口/V | 种类 | 电导率/(S·cm-1) | 文献 |
| 70Li2S-30P2S5 | 5 | glass | 1.58×10-3 | [35] |
| 80Li2S-20P2S5 | 5 | glass-ceramic | 4.3×10-4 | [32] |
| 90Li3PS4·10ZnO | 5 | glass | ﹥10-4 | [31] |
| Li7P2.9S10.85Mo0.01 | 5 | glass-ceramic | 4.8×10-3 | [33] |
| 2.25Li3+0.55P1-0.11S4·0.2LaS2 | 8 | glass-ceramic | 7.41×10-5 | [37] |
| 70Li2S·27P2S5·3P2O5 | 10 | glass-ceramic | 3.0×10-3 | [13] |
| 75Li2S-24P2S5-P2O5 | 10 | glass-ceramic | 8.0×10-4 | [34] |
| Li10GeP2S11.7O0.3 | 10 | crystalline | 1.03×10-2 | [36] |
| 80(0.7Li2S-0.3P2S5)·20LiI | 10 | glass | 5.6×10-4 | [29] |
| 95(0.8Li2S-0.2P2S5)·5LiI | 10 | glass-ceramic | 2.7×10-3 | [30] |
下载: 导出CSV
表 3 含不同掺杂物的硫化物固态电解质的电导率(S·cm-1)
Table 3. Conductivities of sulfide-based solid electrolytes with various doping materials (S·cm-1)
| 组成 | 电导率/(S·cm-1)a | 种类 | 文献 |
| 99(70Li2S·30P2S5)·Li2ZrO3 | 2.85×10-3(1.65×10-3) | glass-ceramic | [38] |
| Li7P2.9S10.85Mo0.01 | 4.8×10-3(2.6×10-3) | glass-ceramic | [33] |
| Li3.05Ge0.05P0.95S4 | 1.2×10-3(9.4×10-4) | glass-ceramic | [39] |
| 98(70Li2S·30P2S5)·2GeS2 | 5.4±0.1×10-3(4.2×10-3) | glass-ceramic | [45] |
| 95(0.8Li2S·0.2P2S5)·5LiI | 2.7×10-3(8.2×10-4) | glass-ceramic | [30] |
| 80(0.7Li2S·0.3P2S5)·20LiI | 5.6×10-4(1.3×10-4) | glass | [29] |
| 90(0.7Li2S·0.3P2S5)·10LiBr | 6.5×10-3(5.2×10-3) | glass-ceramic | [40] |
| 70Li2S·29P2S5·P2S3 | 5.4×10-3(4.2×10-3) | glass-ceramic | [41] |
| 70Li2S·27P2S5·3P2O5 | 4.6×10-3(4.2×10-3) | glass-ceramic | [41] |
| 97(0.78Li2S·0.22P2S5)·3Li3BO3 | 1.03×10-3(6.14×10-4) | glass-ceramic | [42] |
| 30LiI·70(0.07Li2O·0.68Li2S·0.25P2S5) | 1.3×10-3(~5×10-4) | glass | [43] |
| 86.9Li3PS4·13.1LiAlS2 | 6.0×10-4(2×10-4~3×10-4) | glass | [44] |
| 注:a括号里的数值代表未掺杂样品的离子电导率值。 | |||
下载: 导出CSV
表 4 复合正极材料的构成比例
Table 4. Composition ratio of composite cathode materials
| 固态电解质 | 电导率/(S·cm-1) | 活性物质 | 导电剂 | 固态电解质/活性物质/导电剂(质量比) | 放电容量/(mAh·g-1) | 文献 |
| 70Li2S-30P2S5 | 1.58×10-3 | Li2S | Super P+VGCF | 30/60/6.7+3.3 | 1055(1st) | [35] |
| Li7P2.9S10.85Mo0.01 | 4.8×10-3 | S | Super P | 60/30/10 | 1020(1st) | [33] |
| 99(70Li2S·30P2S5)·Li2ZrO3 | 2.85×10-3 | LiCoO2 | 3/8 | 113(1st) | [35] | |
| 30LiI·70(0.07Li2O·0.68Li2S·0.25P2S5) | 1.3×10-3 | LiCoO2/LiNbO3 | 30/70 | 116(30st) | [40] | |
| 97(0.78Li2S·0.22P2S5)·3Li3BO3 | 1.03×10-3 | LiCoO2 | Super P | 19.6/78.4/2 | 149(40st) | [39] |
| Li3.05Ge0.05P0.95S3.9Se0.1 | 1.4×10-3 | LiCoO2 | Super P | 30/20/3 | 62(10st) | [50] |
| 70Li2S·29P2S5·P2S3 | 5.4×10-3 | Li4Ti5O12 | VGCF | 60/40/4 | 140(700st) | [38] |
| Li10GeP2S12 | 1.2×10-2 | LiNi0.5Mn1.5O4/LiNbO3 | Super P | 57.5/38.5/4 | 80(1st) | [51] |
| 78Li2S-22P2S5 | 2.0×10-4 | LiCoO2/Li2CO3 | Super-P | 34/64/2 | 137.4(1st) | [52] |
| 80Li2S-20P2S5 | FePS3 | 107(30st) | [49] | |||
| 78Li2S-22P2S5 | 8.5×10-4 | LiCoO2 | Super P | 59/39/2 | 81(20st) | [48] |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们