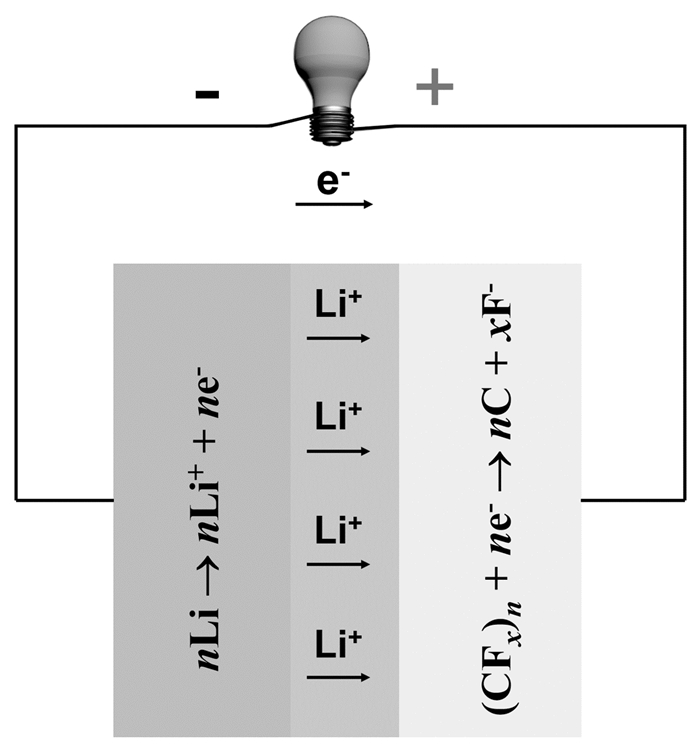
图 1.
锂/氟化碳原电池的工作原理示意图
Figure 1.
Schematic illustration of the discharge mechanism of lithium/carbon fluorides batteries

随着科技的进步和人类需求的提高,以及军事装备向机械化、信息化和数字化方向发展,对各种高性能军用电源的要求越来越高,尤其在卫星、飞船、空间站、深空探测(火星、小行星)登陆器、上面级、轨道转移飞行器等空间应用以及单兵系统、无人机等方面,除了要求各部件重量尽可能轻,以提高有效载荷之外,还需要电源能够大电流放电,提供较高的功率,以满足军事装备的快速响应。
锂/氟化碳(Li/CFx)原电池是首先商品化的一种固体正极锂电池,它的理论质量比能量约为2203Wh/kg[1]。锂/氟化碳原电池具有平稳的放电平台、优异的安全性和贮存性能以及宽的工作温度范围等优势。但是由于正极材料氟化碳的本征导电性差、电极动力学过程缓慢,导致锂/氟化碳原电池在放电过程中产生较大的极化,使锂/氟化碳电池仅限于在低倍率下放电,并在放电初期存在电压滞后;同时,在放电过程中伴随着大量的热耗,影响电池的性能,严重地制约了锂/氟化碳电池的应用。因此,锂/氟化碳电池的应用很长时间以来一直仅限于电子计算机、钟表、照相机以及集成电路存储器和医疗器械上等一些小电流放电的场合。
近年来,航天、军事、钻探等领域飞速发展,对高比能、高安全性、长搁置寿命电能源的需求越来越高,人们又重新把目光放到比能量优异的锂/氟化碳电池上。通过对反应机理认识的不断提高、碳材料的改进以及采用纳米技术来提高锂/氟化碳电池的功率特性,使其有可能更好地应用于各种苛刻的场合。兼具比能量与比功率的锂/氟化碳电池能很好地满足一箭多星、小卫星或太空武器等机动变轨发射、动能拦截弹用电池的需要,具有相当大的竞争优势; 在能源钻探领域,耐高温、高安全性、高倍率特性的锂/氟化碳电池则是采集深井数据的有力保障。
锂/氟化碳原电池以金属锂作为负极,以固体的氟化碳材料作为正极。氟化碳材料是通过氟与碳直接反应生成的插层化合物[2],这类物质虽然在电化学上具有活性,但是在有机电解质中的化学稳定性却很高,因此具有很长的贮存寿命。锂/氟化碳电池在放电过程中,氟化碳材料的C-F键断裂,氟化碳转化为导电的碳,同时放电产物LiF沉积在正极及其导电网络中。锂/氟化碳电池的放电原理至今仍存在一些争议,普遍接受的是,在放电过程中,溶剂化的锂离子进入氟化碳层间,生成三元中间相CLixF或者CFLix:Sy(S为与锂共嵌入的溶剂),这种化合物然后自发分解为C+LiF+溶剂,使开路电压从4.5V降至3.0~3.5 V,同时在反应过程中产生大量的热[3]。电池的简化放电反应如式(1)~(3)所示,工作原理示意图如图 1所示。
|
$ 负极:n \mathrm{Li} \rightarrow n \mathrm{Li}^{+}+n \mathrm{e}^{-} $ |
(1) |
|
$ 正极:\left(\mathrm{CF}_{x}\right)_{n}+n \mathrm{e}^{-} \rightarrow n \mathrm{C}+x \mathrm{F}^{-} $ |
(2) |
|
$ 总反应:n \mathrm{Li}+\left(\mathrm{CF}_{x}\right)_{n} \rightarrow n \mathrm{LiF}+n \mathrm{C} $ |
(3) |
近年来,随着实验设计的完善和各种测试手段的提高,研究者们对锂/氟化碳原电池的放电机理有了更进一步的理解。Zhang等[4]利用三电极体系研究锂/氟化碳电池的放电机理,提出“核壳”模型,即氟化碳材料放电过程中会形成一个“核壳”结构,其中,核为氟化碳材料,壳为产物[产物壳层包括嵌锂态插层中间相GIC(Graphite intercalation compound,石墨插层化合物)、碳和LiF]。也就是说,产物层首先出现在氟化碳表面,然后随着反应的进行,逐渐渗透到材料内部,直至氟化碳材料放电结束。采用固态核磁共振谱可以细致地分析氟化碳材料在放电过程中的非活性区和反应区[5, 6]。
碳材料的氟化是通过打破碳层π键,并和碳原子结合形成键能很高的C-F键的一种过程,生成的氟化碳材料是一种电子电导和离子电导都很低的电极材料,这一特性必然导致氟化碳材料的倍率特性较差,极大地限制了锂/氟化碳电池的功率特性。此外,氟化碳在放电过程中生成的放电产物LiF是电子绝缘和离子绝缘的物质,随着放电的进行,LiF不断覆盖在正极电极上,影响电极的功率特性。
根据目前的研究,针对锂/氟化碳原电池倍率特性的主要优化设计措施有:通过改变氟化碳材料的前驱体制备不同的氟化碳材料、氟化方法的改进、氟化碳材料表面修饰和氟化碳正极结构优化等。
目前已经商业化应用的氟化碳材料为氟化石墨,但是石墨的片状结构不利于锂离子电化学反应进行,特别是高倍率放电条件下。现在随着碳材料科学与纳米技术的发展,研究者在材料改善方面进行了不少尝试。采用新型碳材料作为前驱体是改善氟化碳材料在锂电池中的倍率性能的一种普遍的方法。例如,采用纳米碳管[7~9]、纳米碳纤维[10, 11]、介孔碳材料[12]、富勒烯[13~16]、石墨烯[17]等作为前驱体进行氟化,相比传统的氟化石墨,能够在一定程度上改善锂/氟化碳原电池的倍率特性。
Hamwi等[7]最早通过氟气在不同的温度下制备氟化多壁碳纳米管(F-MWCNTs)。结果表明,当温度高于300℃时,通过透射电镜观察到碳管的表面出现皱层,但管状结构保持良好;当温度高于400℃时,氟化会使MWCNTs表面出现微小裂纹,这为氟原子渗入MWCNTs内部从而提高氟碳比提供了路径。由于MWCNTs是由围绕纳米管中心的多层石墨层构成,因此制备F-MWCNTs的反应温度明显高于制备氟化单壁碳纳米管(F-SWNTs)的反应温度[8]。一般而言,接近400℃才能在MWCNTs表面形成C-F键。采用F2、HF和IF5的混合气体作为氟源(低温氟化),可在室温下制备F-MWCNTs,虽然表面存在大量的皱褶,但MWCNTs的管状结构得以保持,这样制备的产品具有一定的导电能力。同时,氟原子仅分布在MWCNTs外层,这为提高氟化碳材料导电性从而提高锂/氟化碳原电池的倍率性能提供了新的思路和技术条件。
通过对不同孔径的MWCNTs进行氟化[9],控制氟碳比在1左右,所制备的F-MWCNTs可以在4C下进行放电,孔径大于50nm的材料可以在5C下放电,最大功率密度可达7114.1W/kg,能量密度为1923Wh/kg(基于材料计算)。MWCNTs自身的导电网络和一维的快速电子传输速率是F-MWCNTs材料具有优异倍率性能的主要原因。而孔径较大的材料可以在放电过程中避免F-MWCNTs被放电产物LiF堆积,造成电导率下降,从而具有更优异的倍率性能。
通过对不同氟化度的氟化碳纤维的倍率性能进行比较得出,氟碳比为0.76的氟化碳纤维具有优异的倍率性能,可在6C下进行放电,最高功率密度可达8057W/kg(按照材料计算)[10]。控制氟碳比,使得在纳米尺度上氟化和非氟化部分共存,中心未氟化的、具有石墨结构的部分作为电子快速运输部分,降低因C-F键引起的欧姆降。氟化碳纤维中具有10%的石墨型碳即可改善其电导率,减少高倍率下的电压极化。
使用介孔碳材料作为前驱体,低温下制备出的氟化介孔碳材料具有较窄介孔分布(6~11nm),比表面积高达852m2/g,其分级式孔径和孔分布使得氟化介孔碳材料兼具高比能量和高功率特性[12]。在5C(3136mA/g,3994mA/cm2)倍率下,比容量仍有515mAh/g,电压平台为2.75V,高于商业化氟化石墨的放电平台。
相比其他碳材料,富勒烯(C60或C70)对氟化温度要求较低,在室温下就能与氟气反应生成较高氟化度的氟化富勒烯(如C60F46~54[13]或C70F52~56[14])[15]。更高的氟化度需要在近300℃的温度下获得,且结晶度较低氟化度的富勒烯高很多。300℃以下,C60和C70仍能维持笼状结构,300℃以上则出现破裂的趋势。虽然氟化富勒烯的合成工艺有了重大的突破,但是其易溶解于常用的有机溶剂电解液,严重影响了它的实际应用。不过,如果将氟化富勒烯应用于固体电解质体系中,就能比普通氟化碳材料在小电流下达到更高的放电电压,而且没有容量损失[16]。
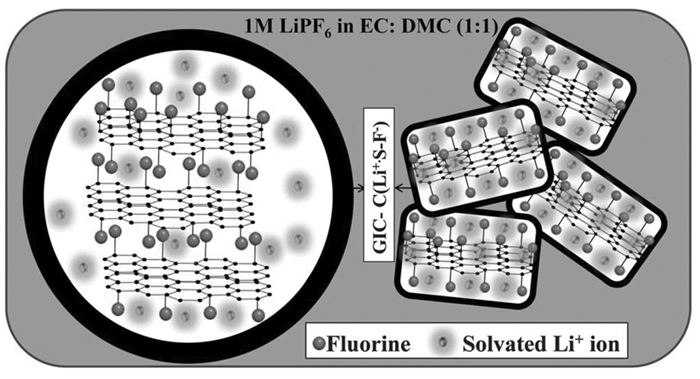
石墨烯具有优异的本征电子传导性,氟化后,由于C-F键的形成,会存在大量的缺陷,将加速锂离子的传导,使得氟化石墨烯具有优异的电化学性能。可采用制备氧化石墨烯和石墨烯的方法制备氟化氧化石墨烯,并还原制备氟化石墨烯(FG)[17]。FG相比氟化石墨,呈现出层状纳米片结构,具有良好的倍率特性。在1000mA/g的电流密度下,比容量为572mAh/g。在FG中,纳米片层状结构使得GIC层很薄,加速溶剂化的锂离子与氟的反应,同时使反应更彻底(如图 2所示)。此外,FG材料中大量的氟都存在于材料表面,不仅加快了反应速率,还提高了比容量。
传统的氟化碳制备方法主要有两种:高温制备法[18]和低温制备法[19]。高温氟化碳氟化程度高、氟碳比可控度高,然而C-C之间以sp3杂化为主,C-F共价键键能较高,导电性较差,导致倍率性能较差。低温氟化碳由碳源在低温下被Lewis酸氧化形成[20~22]。C-C之间保留了较多的sp2杂化结构,F夹杂在石墨片层中间[23]。这样产生的氟化碳以离子态或半离子态形式存在,具有非常好的电子电导性,颜色呈黑色,这与高温氟化石墨白至浅灰色及绝缘性有着明显的差异。传统的商用锂/氟化碳原电池均采用高温氟化碳材料,氟碳比≥1,优先考虑了以高氟化度带来的高能量密度。低温氟化石墨在5C下放电比容量约430mAh/g,而高温氟化石墨仅可以3C放电,并且在低倍率下低温氟化石墨的放电平台高出高温氟化石墨约0.5V[24]。但是与其他改性方法相比,低温氟化后残留的IF5等催化剂会影响锂/氟化碳原电池的储存寿命[25]。
此外,可以通过控制高温氟化法的氟化温度和氟化时间进行部分氟化。Chamssedine等[26]以碳纤维作为前驱体,通过控制反应的温度,对碳纤维表面进行氟化,产物保留了碳纤维管内纳米级的未氟化碳原子,从而增加氟化碳材料的导电性,大大地提高了锂/氟化碳电池的功率密度。部分氟化法已由美国加州理工大学及法国国家科学研究中心共同开发授权,被美国Contour公司采用,可用于制备兼具高比能量及高功率的空间应用氟化碳电池,在大倍率放电下,能量保持率比传统的氟化碳电池提高了10倍。
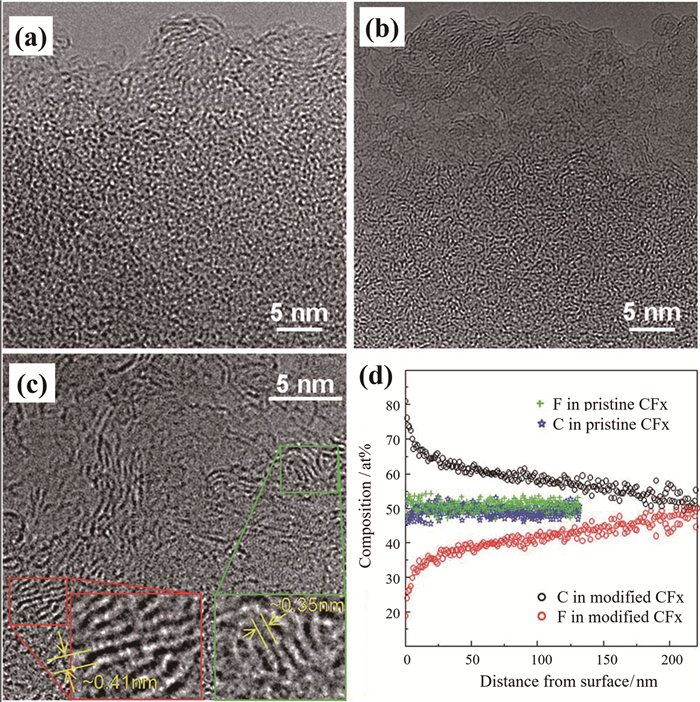
对氟化碳材料的表面进行修饰也是一种较常用的方法,例如使用碳热包覆法[27, 28]、碳包覆[29]、水热处理等[30, 31]。碳热包覆方法是将氟化碳、导电剂、氟化粘结剂压成片,在低于氟化碳分解的温度下进行热处理。利用氟化聚合物粘结剂的粘结性及生成的HF作为催化剂,最终形成半氟化的碳,改善其倍率性能。同样,热处理方法与碳热包覆法类似,是在低于氟化碳分解温度下对氟化碳进行热处理,形成部分半氟化的碳,以提高倍率特性。将商用的氟化石墨在180℃下进行水热处理[30, 31],制备出具有氟元素梯度的氟化碳材料。图 3为制备的氟化碳梯度材料的透射电镜图及元素分布。材料表面的氟元素含量低,呈多孔石墨烯状,电导率增加,加速锂离子的传输;材料内部的氟元素含量较高,使材料仍具有高的比容量。该氟化碳材料在30C下比容量为500mAh/g,功率密度高达44800W/kg(按材料计算)。
Li等[32]以MWNTs代替乙炔黑作为导电剂,利用其构建三维导电网络,并利用其本身优异的电导率,改善倍率性能,1C下放电比容量达到712mAh/g。Meduri等[33]将氟化碳、Ag2V4O11和石墨烯混合球磨。在球磨过程中,一方面减少了CF2、CF3等表面非活性基团,另一方面石墨烯和还原的Ag颗粒均匀分散在氟化碳上,形成良好的连续导电网络,5C下放电比容量达到462mAh/g,该微型电池可用于水下发射器。Groult等[34]在氟化碳表面沉积一层约10nm的聚吡咯(PPy),使锂/氟化碳电池的功率密度提高了近4倍,达到了5235W/kg(4C)。
Yang等[35]采用石墨烯纸及石墨烯/贵金属(Ag,Au)复合纸作为集流体,在有效提高锂/氟化碳电池功率密度的同时,兼顾了体系的能量密度。以石墨烯纸为集流体的锂/氟化碳电池在5C倍率下,能量密度可达1350Wh/kg,功率密度可达8000W/kg,同时可以缓解放电过程中因体积膨胀造成的接触不良。此外,其作为一种轻质柔性的电极,扩展了锂/氟化碳电池的应用范围。
Reddy等[36]通过球磨减小氟化石墨的颗粒度,加速反应动力学,改善了材料的倍率特性,在6C下仍保持了40%的比容量。球磨后的氟化石墨材料的能量密度为800Wh/kg,功率密度达到9860W/kg。加入氧化物正极材料可减小电压滞后和提高倍率特性等[37, 38]。将二氧化锰(MnO2)与CFx混合,在5C下功率密度达到6599W/kg,同时能量密度维持在1814Wh/kg[37]。同样,在提供相同的脉冲能量时,Li/(CFx-MnO2)电池(BA-5790)相比商业化的Li/SO2电池(BA-5590)具有更轻的质量和更小的体积[38]。法国SAFT公司制备的LH33550 D型电池即采用MnO2与CFx的混合体系,用MnO2弥补氟化碳电导率低的缺点,提高了CFx电池的倍率性能。在电极界面方面的研究主要集中在电极/电解液界面的改进,使用对LiF溶解度更大的电解液[11],或者在电解液中加入可以溶解LiF的添加剂[39, 40],改善放电过程中正极电导率来提高倍率特性等。
在高能量的能源领域中,锂/氟化碳原电池有很好的应用前景,提高其功率特性可以扩展其应用范围。本文探讨了提高锂/氟化碳电池功率特性的方法,可以概括为提高电池中的离子电导和电子传导。但是在实际应用情况下,应从电池体系中的正极、电解液、隔膜、界面等各方面进行全面优化设计,相信在不久的将来,兼具高能量密度和功率密度的锂/氟化碳原电池可以在军事及民用领域得到更广泛的应用。
H Touhara, F Okino. Carbon, 2000, 28:241~267.
G G Amatucci, N Pereira. J. Fluorine Chem., 2007, 128:243~262. doi: 10.1016/j.jfluchem.2006.11.016
R Hagiwara, T Nakajima, N Watanabe. J. Electrochem. Soc., 1988, 135:2128~2133. doi: 10.1149/1.2096228
S S Zhang, D Foster, J Wolfenstine et al. J. Power Sources, 2009, 187:233~237. doi: 10.1016/j.jpowsour.2008.10.076
N D Leifer, V S Johnson, R Ben-Ari et al. J. Electrochem. Soc., 2010, 157:A148~A154. doi: 10.1149/1.3267042
J H S R DeSilva, R Vazquez, P E Stallworth et al. J. Power Sources, 2011, 196:5659~5666. doi: 10.1016/j.jpowsour.2011.02.036
A Hamwi, H Alvergnat, S Bonnamy et al. Carbon, 1997, 35:723~728. doi: 10.1016/S0008-6223(97)00013-4
E T Mickelson, C B Huffman, A G Rinzlera et al. Chem. Phys. Lett., 1988, 296:188~194.
Y Li, Y Feng, W Feng. Electrochim. Acta, 2013, 107:343~349. doi: 10.1016/j.electacta.2013.06.086
R Yazami, A Hamwi, K Guérin et al. Electrochem. Commun., 2007, 9:1850~1855. doi: 10.1016/j.elecom.2007.04.013
Y Ahmad, K Guérin, M Dubois et al. Electrochim. Acta, 2013, 114:142~151. doi: 10.1016/j.electacta.2013.09.140
P F Fulvio, S S Brown, J Adcock et al. Chem. Mater., 2011, 23:4420~4427. doi: 10.1021/cm2012395
Y Matsuo, T Nakajima. Electrochim. Acta, 1996, 41:15~19. doi: 10.1016/0013-4686(95)00288-P
D Claves, A J Giraudet, A Hamwi et al. J. Phys. Chem. B, 2001, 105:1739~1742. doi: 10.1021/jp0026336
A Hamwi. J. Phys. Chem. Solids, 1996, 57:677~688. doi: 10.1016/0022-3697(95)00332-0
N Liu, H Touhara, F Okino et al. J. Electrochem. Soc., 1996, 143:2267~2272. doi: 10.1149/1.1836992
D Damien, P M Sudeep, T N Narayanan et al. RSC Adv., 2013, 3:25702~25706. doi: 10.1039/c3ra45377d
H Fujimoto, A Mabuchi. Carbon, 1992, 30(2):851~857.
R Hagiwara, M Lerner, N Bartle. J. Electrochem. Soc., 1988, 135:2393~2394.
C Delabarre, K Guérin, M Dubois et al. J. Fluorine Chem., 2005, 126:1078~1087. doi: 10.1016/j.jfluchem.2005.03.019
A S Nazarov, V G Makotchenko. Inorg. Mater., 2002, 38(3):278~282. doi: 10.1023/A:1014783119281
V Gupta, T Nakajima, Y Ohzawa et al. J. Fluorine Chem., 2003, 120:143~150. doi: 10.1016/S0022-1139(02)00323-8
P Lam, R Yazami. J. Power Sources, 2006, 153:354~359. doi: 10.1016/j.jpowsour.2005.05.022
R Yazami, P Hany, P Masset et al. Mol. Cryst. Liq. Cryst., 1998, 310:397~402. doi: 10.1080/10587259808045368
J Giraudeta, C Delabarrea, K Guérina et al. J. Power Sources, 2006, 158:1365~1372. doi: 10.1016/j.jpowsour.2005.10.020
F Chamssedine, M Dubois, K Guérin et al. Chem. Mater., 2007, 19:161~172. doi: 10.1021/cm061731m
S S Zhang, D Foster, J Read. J. Power Sources, 2009, 188:601~605. doi: 10.1016/j.jpowsour.2008.12.007
S S Zhang, D Foster, J Read. J. Power Sources, 2009, 191:648~652. doi: 10.1016/j.jpowsour.2009.02.007
L Zhu, Y Pan, L Li et al. J. Electrochem. Sci., 2016, 11:14~22.
Y Dai, S Cai, L Wu et al. J. Mater. Chem. A, 2014, 2:20896~20901. doi: 10.1039/C4TA05492J
Y Dai, Y Fang, S Cai et al. J. Electrochem. Soc., 2017, 164(2):A1~A7.
Y Li, Y Chen, W Feng et al. J. Power Sources, 2011, 196:2246~2250. doi: 10.1016/j.jpowsour.2010.10.005
P Meduri, H Chen, X Chen et al. Electrochem. Commun., 2011, 13:1344~1348.
H Groult, C M Julien, A Bahloul et al. Electrochem. Commun., 2011, 13:1074~1076. doi: 10.1016/j.elecom.2011.06.038
W Yang, Y Dai, S Cai et al. J. Power Sources, 2014, 255:37~42. doi: 10.1016/j.jpowsour.2013.12.122
M A Reddy, B Breitung, M Fichtner. ACS Appl. Mater. Interf., 2013, 5:11207~11211. doi: 10.1021/am403438m
Y Li, W Feng. J. Power Sources, 2015, 274:1292~1299. doi: 10.1016/j.jpowsour.2014.10.150
P H Smith, R B Sepe Jr, K G Waterman et al. J. Power Sources, 2016, 327:495~506. doi: 10.1016/j.jpowsour.2016.07.035
N G Nair, M Blanco, W West et al. J. Phys. Chem. A, 2009, 113:5918~5926. doi: 10.1021/jp901952t
L F Li, H S Lee, H Li et al. Electrochem. Commun., 2009, 11:2296~2299. doi: 10.1016/j.elecom.2009.10.015

图 1 锂/氟化碳原电池的工作原理示意图
Figure 1 Schematic illustration of the discharge mechanism of lithium/carbon fluorides batteries

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

