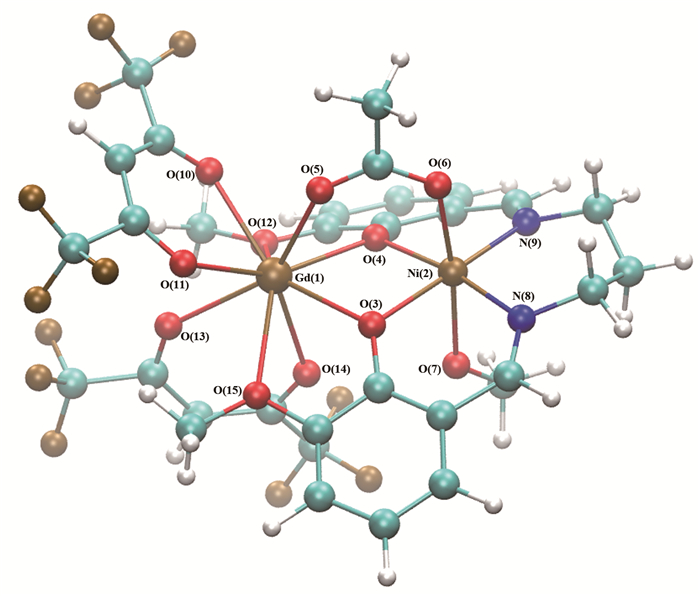
图 1.
配合物[Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]分子结构
Figure 1.
Molecular structure of complex [Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]

自1968年Dutt等[1]第一次合成了以席夫碱为配体的稀土配合物以来,含GdⅢ和NiⅡ等过渡金属离子的3d-4f配合物,因其有大的基态自旋和负的各向异性,在单分子磁体及磁弛豫分子材料等领域受到广泛关注[1~6]。稀土离子GdⅢ具有高自旋基态和磁各向同性,其离子间的磁交换作用较弱,而3d顺磁性过渡金属离子(如CuⅡ、NiⅡ、CoⅡ等)间的磁交换作用通常比稀土离子的要强。将3d过渡金属离子和4f稀土离子结合在同一配合物中,稀土离子与3d过渡金属离子间通过桥联配体相互连接,产生较强的磁耦合作用,使得新生成的配合物具有较大的基态自旋。改变配体的类型和结构,通过调节桥联配位原子的桥联方式或桥联原子数目,可以合成性能千差万别的3d-4f多功能分子基磁体。为此,前人设计不同的多齿配体,合成了大量磁学性质和结构各异的3d-4f分子基磁体[7~20],并开展了部分理论研究[21~26]。其中氧桥联GdNi双核配合物是研究较多的一类3d-4f分子基磁体,实验研究表明,大多数氧桥联GdNi配合物顺磁中心GdⅢ和NiⅡ之间存在铁磁性相互作用,其磁耦合常数随桥联氧原子与顺磁中心形成的夹角(∠GdONi)的增大而增大[21, 23~25]。Singh等[21]采用密度泛函理论结合对称性破损态方法(DFT-BS)研究双核NiⅡGdⅢ配合物顺磁中心间磁交换作用和磁性与结构关系,结果表明,顺磁中心GdⅢ和NiⅡ之间磁耦合常数随∠GdONi键角的增加而增大,为磁学性质与结构关系的探究提供了较好的基础,但对磁学性质与结构关系的深入探究仍有不足。本文以3d过渡金属离子NiⅡ、4f稀土离子GdⅢ、六氟乙酰丙酮为主要配体、醋酸和甲醇为辅助配体合成的3d-4f双核配合物为研究对象,探究其磁学性质与结构的关系,可为3d-4f分子基磁体可控合成提供一定的理论参考。
GdⅢNiⅡ双核配合物顺磁中心GdⅢ和NiⅡ之间的相互作用可用Heisenberg-Dirac-Van Vleck(HDVV)哈密顿算符表示:
|
$ \hat H{\rm{ = }} - 2{J_{{\rm{GdNi}}}}{\hat S_{{\rm{Gd}}}} \cdot {\hat S_{{\rm{Ni}}}} $ |
(1) |
其中,JGdNi表示两个顺磁中心GdⅢ和NiⅡ之间磁耦合常数,它的符号和大小表示顺磁中心GdⅢ和NiⅡ之间相互作用的类型和强弱[10]。JGdNi > 0,顺磁中心GdⅢ和NiⅡ之间是铁磁性相互作用,JGdNi值越大,铁磁性相互作用越强;JGdNi < 0,顺磁中心GdⅢ和NiⅡ之间是反铁磁性相互作用,JGdNi值越小,反铁磁性相互作用越强[10]。
采用DFT-BS方法,计算高自旋态|7/2, 1>的能量(EHS)和对称性破损态|7/2, -1>的能量(EBS),其磁耦合常数J值使用以下公式计算:
|
$ J = - \frac{{{E_{{\rm{HS}}}} - {E_{{\rm{BS}}}}}}{{S_{{\rm{HS}}}^2 - S_{{\rm{BS}}}^2}} $ |
(2) |
其中SHS2和SBS2表示HS态和BS态自旋平方算符的期望值。由于J值对配合物的结构参数很敏感,在考察计算条件时,为确保计算时配合物所处环境与其真实环境一致,配合物结构直接来自单晶衍射数据,未做优化[7]。高自旋态(HS态) |7/2, 1>为十重态,对称性破损态(BS态) |7/2, -1>为六重态。在计算BS态时,采用FlipSpin和FinalMs关键词通过交换HS顺磁中心NiⅡ的α和β自旋密度实现。由于BS态是通过交换HS态波函数中顺磁中心NiⅡ的α和β自旋密度实现,没有改变配合物分子的核构型,在计算HS态和BS态的能量时,配合物结构均取自实验得到的单晶衍射数据,未做改动[27]。所有计算使用ORCA4.0.1软件[28~31]在贵州工程应用技术学院计算化学实验室正睿I2TS2-8898HV服务器上完成[30, 31],配合物结构及相关图形用VMD 1.9.3[32]显示。
磁耦合常数J值对分子结构参数非常敏感,分子结构参数(如键长、键角)的变化会引起J值的变化,甚至会出现磁学性质的改变。所以GdⅢNiⅡ双核配合物模型中所有的原子坐标均来源于X-射线衍射所得的单晶结构[6],如图 1所示。
采用DFT-BS方法,相对论效应使用DKH2方法,稀土元素Gd选用Neese等[33]提出的全电子相对论基组SARC2-DKH-QZVP,其他元素选择TZVP基组,数值积分精度参数为6.0。选取广义梯度近似(GGA)方法(LDA、VWN3、VWN5、BLYP、XLYP、BP86)、杂化密度泛函(HDFT)方法(B3LYP、B3P86、X3LYP、O3LYP、PBE0、mPW1LYP、mPW1PW)、含动能密度的meta-GGA泛函方法(TPSS、TPSS0、TPSSh、M06、M06-L、M06-2X)和含二阶微扰校正的双混合泛函方法(B2PLYP、mPW2PLYP、PWPB95)计算配合物顺磁中心GdⅢ和NiⅡ之间的磁耦合常数Jcalc值,计算结果如表 1所示。由表可知,四类方法计算的磁耦合常数Jcacl值均大于零,说明采用四类方法在TZVP基组(Gd为SARC2-DKH-QZVP)下计算的磁学性质与实验测量值一致。GGA泛函计算的结果在7.39~54.30 cm-1之间,极差为46.91cm-1。其中BP86泛函计算值54.30cm-1与实验值1.10cm-1差值最大。杂化密度泛函计算的Jcacl值1.04~1.68 cm-1之间,极差为0.64cm-1,其中X3LYP泛函计算值1.13cm-1与实验值1.10cm-1最接近。meta-GGA泛函计算的磁耦合常数Jcacl值在0.79~1.99 cm-1之间,极差为1.20cm-1,TPSS和TPSS0泛函计算的1.12cm-1与实验值1.10cm-1最吻合。双混合泛函计算的磁耦合常数Jcacl值在0.55~0.62cm-1之间,极差为0.07cm-1,其计算的磁耦合常数Jcacl值约为实验值1.10cm-1的一半。在计算配合物磁耦合常数中最常用的B3LYP和BP86计算的磁耦合常数Jcacl值分别为1.18和54.30 cm-1,B3LYP泛函计算的结果与实验值1.10cm-1较吻合,与meta-GGA泛函计算的结果也相近。所以,选用B3LYP方法研究GdⅢNiⅡ双核配合物磁学性质。
 下载:
导出CSV
下载:
导出CSV
| 泛函 | EHS/eV | EBS/eV | SHS2 | SBS2 | J/cm-1 |
| LDA | -438845.7838 | -438845.7709 | 24.8425 | 10.8131 | 7.39 |
| VWN3 | -439129.5037 | -439129.4907 | 24.8394 | 10.8047 | 7.47 |
| VWN5 | -438845.7838 | -438845.7709 | 24.8425 | 10.8131 | 7.39 |
| BLYP | -439833.5984 | -439833.5906 | 24.8133 | 10.7845 | 4.48 |
| XLYP | -439929.0020 | -439928.9946 | 24.8111 | 10.7820 | 4.28 |
| BP86 | -439872.0800 | -439871.9861 | 25.1168 | 11.1705 | 54.30 |
| TPSS | -439808.5077 | -439808.5058 | 24.7635 | 10.7632 | 1.12 |
| TPSSh | -439824.9913 | -439824.9886 | 24.7663 | 10.7654 | 1.59 |
| TPSS0 | -439808.5077 | -439808.5058 | 24.7635 | 10.7632 | 1.12 |
| M06 | -439793.4064 | -439793.4048 | 24.7687 | 10.7684 | 0.94 |
| M06-L | -439815.5809 | -439815.5775 | 24.7849 | 10.7807 | 1.99 |
| M06-2X | -439830.6805 | -439830.6791 | 24.7666 | 10.7664 | 0.79 |
| B2PLYP | -439634.3555 | -439634.3545 | 24.7627 | 10.7626 | 0.57 |
| mPW2PLYP | -439645.5448 | -439645.5438 | 24.7627 | 10.7625 | 0.55 |
| PWPB95 | -439704.1563 | -439704.1552 | 24.7622 | 10.7621 | 0.62 |
| B3LYP | -439786.7372 | -439786.7352 | 24.7638 | 10.7634 | 1.18 |
| B3P86 | -439825.1466 | -439825.1444 | 24.7648 | 10.7643 | 1.29 |
| X3LYP | -439762.2324 | -439762.2304 | 24.7636 | 10.7632 | 1.13 |
| O3LYP | -439729.1774 | -439729.1745 | 24.7731 | 10.7717 | 1.68 |
| PBE0 | -439722.6541 | -439722.6521 | 24.7641 | 10.7638 | 1.16 |
| mPW1LYP | -439810.6793 | -439810.6774 | 24.7630 | 10.7626 | 1.04 |
基组大小对计算结果也有一定影响,因此选择B3LYP方法,在相对论效应为DKH2、数值积分精度参数为6.0的条件下考察基组对磁耦合常数计算的影响,其结果如表 2所示。从表 2可以看出,在B3LYP方法下计算的结果与基组的依赖性较小。Gd原子选择SARC2-DKH-QZVP、SARC-DKH-TZVPP、SARC-DKH-TZVP,其他原子用QZVP、TZVP、TZV基组,计算得到的磁耦合常数Jcalc值的极差为0.03cm-1,与实验值1.10cm-1都比较接近。但基组太小,计算结果的可信度不高,所以选择磁耦常数计算时常用的B3LYP方法,在较大的TZVP(Gd为SARC2-DKH-QZVP)基组下研究GdⅢNiⅡ双核配合物的磁学性质。
下载:
导出CSV
| 基组 | EHS/eV | EBS/eV | SHS2 | SBS2 | Jcalc/cm-1 |
| Gd(SARC2-DKH-QZVP) 其他原子(QZVP) |
-439828.5262 | -439828.5242 | 24.7636 | 10.7631 | 1.15 |
| Gd(SARC2-DKH-QZVP) 其他原子(TZVP) |
-439786.7372 | -439786.7352 | 24.7638 | 10.7634 | 1.18 |
| Gd(SARC-DKH-TZVPP) 其他原子(TZVP) |
-439753.0795 | -439753.0774 | 24.7638 | 10.7633 | 1.18 |
| Gd(SARC-DKH-TZVP) 其他原子(TZVP) |
-439753.0795 | -439753.0774 | 24.7638 | 10.7633 | 1.18 |
| Gd(SARC-DKH-TZVP) 其他原子(TZV) |
-439727.9009 | -439727.8989 | 24.7641 | 10.7636 | 1.16 |
为考察其所选择计算条件的可靠性,选择7个结构不同的GdⅢNiⅡ双核配合物,计算磁耦合常数Jcalc值,通过与实验测量值Jexp比较,衡量计算条件的可靠性,其结果如表 3所示。从表 3可以看出,在B3LYP/TZVP(Gd为SARC2-DKH-QZVP)水平下采用DFT-BS方法计算GdⅢNiⅡ双核配合物的磁学性质得到的结果与实验测量的磁学性质一致。所以选择该条件研究GdⅢNiⅡ双核配合物磁学性质是适合的,可用于预测新合成的GdⅢNiⅡ双核配合物的磁学性质。
下载:
导出CSV
| Complexes | EHS/eV | EBS/eV | SHS2 | SBS2 | Jcacl/cm-1 | Jexp/cm-1 |
| [(C21H24N2O4)Ni(H2O)2Gd(NO3)3] | -408402.5386 | -408402.5362 | 24.7607 | 10.7604 | 1.34 | 1.8[35] |
| [Ni(CH3CN)2(valpn)Gd(NO3)3]3·CH3CN | -409316.1196 | -409316.1171 | 24.7620 | 10.7616 | 1.46 | 1.15[20] |
| [(NiL)Gd(hfac)2(EtOH)]·1.5EtOH | -441419.2514 | -441419.2508 | 24.7619 | 10.7615 | 0.33 | 0.34[2] |
| [Ni(HL)(H2O)(tfa)Gd(hfac)2] | -448879.4516 | -448879.4493 | 24.7611 | 10.7607 | 1.32 | 1.83[7] |
| [Ni(valpan)(MeOH)(ac)Ln(hfac)2] | -439727.9010 | -439727.8989 | 24.7641 | 10.7636 | 1.16 | 1.10[6] |
| [Ni(μ-L)(μ-Ac)Gd(NO3)2] | -408668.1892 | -408668.1882 | 24.7599 | 10.7593 | 0.55 | 0.69[18] |
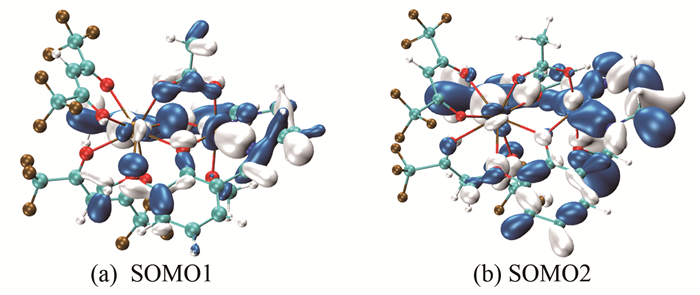
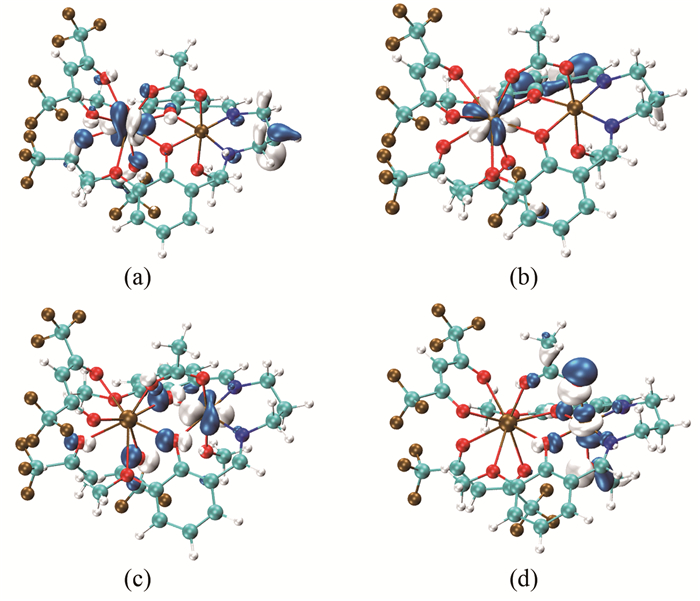
在分子磁学理论研究中,将定域在顺磁中心的轨道称为局域磁轨道,高自旋态的单占据磁轨道(SOMOs)成为分子磁轨道,分析分子磁轨道信息有助于理解配合物顺磁中心间的交换作用[30, 31, 34~37]。
由图 2的分子磁轨道和图 3的局域磁轨道可以看出,配合物[Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]顺磁中心GdⅢ的未成对电子主要定域在4f轨道上,而顺磁中心NiⅡ的未成对电子主要集中在3dz2和3dx2-y2轨道上,且两个顺磁中心GdⅢ、NiⅡ与两个桥联原子(O(3)、O(4))和醋酸桥氧原子(O(5)、O(6))之间存在较强的轨道相互作用。磁轨道的主要贡献来源于顺磁中心NiⅡ的3dz2和3dx2-y2、酚氧桥联配体中氧原子的2pz轨道、醋酸桥中氧原子的2pz轨道和顺磁中心GdⅢ的4fxyz、4fz2x轨道。
对配合物[Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]进行自旋密度分析,可以帮助理解顺磁中心间相互作用机理。若顺磁中心与周围配位原子自旋密度符号相同,则为自旋离域机理;若自旋密度的符号相反,则为自旋极化机理[34, 35~37]。
根据Mulliken布居分析,在B3LYP/TZVP(Gd为SARC2-DKH-QZVP)水平下计算了配合物[Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]HS态和BS态的自旋密度,如图 4所示,其中蓝色和白色分别表示α和β自旋。
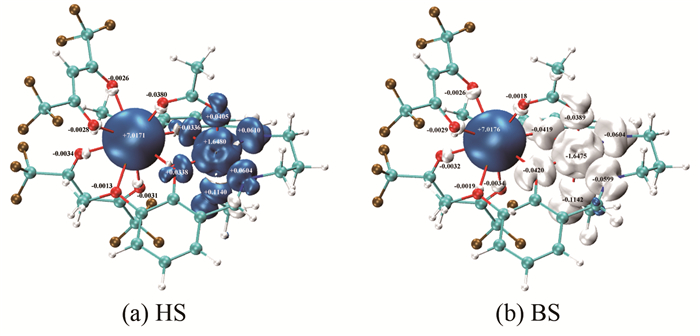
从图 4可知,无论HS态还是BS态,顺磁中心NiⅡ离子和周围配位原子N和O间的自旋密度符号都相同,表明顺磁中心NiⅡ离子主要是自旋离域作用[36]。无论HS态还是BS态,顺磁中心GdⅢ离子和周围配位原子O间的自旋密度符号都相反,表明顺磁中心GdⅢ离子主要是自旋极化作用[36]。在BS态NiⅡ离子的自旋电子(-1.6475e)明显地离域到周围配体上,包括桥联配体O(3),O(4)(-0.0420e,-0.0419e);端基配体N(8),N(9)(-0.0599e,-0.0604e);直接与Ni配位的醋酸根离子和甲醇中的氧,即O(6)(-0.0389e)和O(7)(-0.1142e)。而GdⅢ离子的自旋电子(7.0176e)主要是局域的,自旋密度呈球形分布,并诱导周围配位原子(六氟乙酰丙酮中的O(10)、O(11)、O(12)、O(13)、O(14)、O(15))自旋极化,这种自旋极化的结果使周围配位原子的自旋密度符号与Gd的自旋密度符号相反。从BS态自旋电子在各原子轨道上的分布中可以看到(如表 4所示),Ni的自旋电子主要是3d电子-1.6666e(正号表示α自旋,负号表示β自旋),4s和4p上只有0.0024e,0.00167e。而Gd的自旋电子主要是4f电子6.9469e,也有少量的5d,6s,6p(0.0074e,0.0125e,0.0508e)自旋电子。由此可见,桥联配位原子O(3)、O(4)受到顺磁中心GdⅢ自旋极化和顺磁中心NiⅡ自旋离域的共同作用。桥联配位原子O(3)、O(4),无论在HS态(图 3(a))和BS态(图 3(b))中都与NiⅡ的自旋密度符号相同,这说明NiⅡ的自旋离域作用大于GdⅢ的自旋极化作用对桥联氧原子的影响。
下载:
导出CSV
| 原子 | 原子轨道 | |||
| s | p | d | f | |
| Gd(1) | 0.0074 | 0.0125 | 0.0508 | 6.9469 |
| Ni(2) | 0.0024 | 0.0167 | -1.6666 | |
| O(3) | -0.0055 | -0.0364 | ||
| O(4) | -0.0058 | -0.0360 | ||
| O(5) | -0.0003 | -0.0016 | ||
| O(6) | -0.0042 | -0.0347 | ||
| O(7) | -0.0038 | -0.1102 | ||
| N(8) | -0.0173 | -0.0426 | ||
| N(9) | -0.0169 | -0.0435 | ||
| O(10) | -0.0004 | -0.0025 | ||
| O(11) | -0.0004 | -0.0023 | ||
| O(12) | -0.0002 | -0.0014 | ||
| O(13) | -0.0003 | -0.0030 | ||
| O(14) | -0.0003 | -0.0031 | ||
| O(15) | -0.0002 | -0.0017 | ||
在氧桥联GdNi双核配合物中,配合物磁耦合常数J值随桥联氧原子与两个顺磁中心形成的平均键角∠GdONi (θ)增大而增大[18, 21],当∠GdONi减小时,顺磁中心GdⅢ、NiⅡ之间的磁耦合作用可由铁磁性相互作用转变为反铁磁性相互作用,但未作更深入的探讨。以含醋酸氧桥和酚氧桥联配体的[Ni(HL)(H2O)(tfa)Gd(hfac)2]配合物为模型[7],固定其他原子不动,通过改变键角的大小,计算其顺磁中心GdⅢ、NiⅡ之间的磁耦合常数(Jcalc),探究键角对配合物磁学性质的影响,结果如图 5所示。
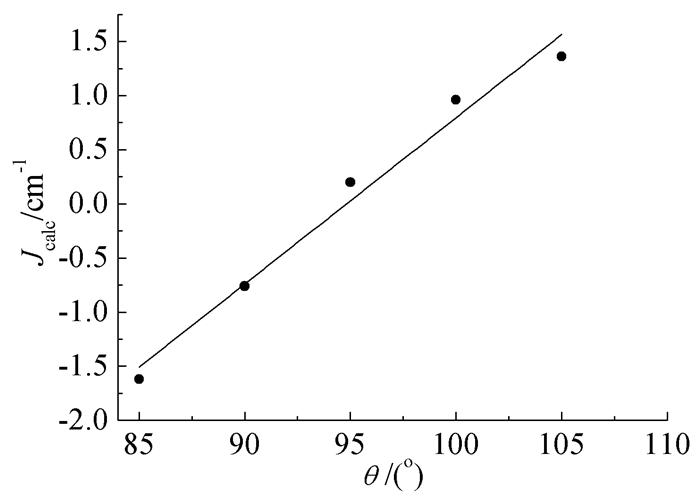
从图 5可知,在氧桥联GdNi双核配合物中,顺磁中心GdⅢ和NiⅡ之间磁耦合常数随键角∠GdONi增大而增大,在85°~105°的范围内,线性关系较好。根据计算得到的磁耦合常数拟合出的方程Jcalc=0.1536θ-14.564,可以推测当键角θ < 94.82°时,顺磁中心GdⅢ和NiⅡ之间由铁磁性相互作用向反铁磁性相互作用转变。
Illas等[38]、Ruiz等[39]证明了顺磁中心HS态和BS态自旋密度的平方差也可以衡量磁耦合作用强弱。磁耦合常数与自旋密度的平方差的关系可以用式(3)表示:
|
$ {J_{{\rm{AF}}}} \propto \rho _{{\rm{HS}}}^2 - \rho _{{\rm{BS}}}^2 $ |
(3) |
顺磁中心GdⅢ和NiⅡ之间磁耦合常数J可表达为铁磁性相互作用贡献(JF)和反铁磁性相互作用贡献(JAF)的总和,即:
|
$ J = {J_{\rm{F}}} + {J_{{\rm{AF}}}} $ |
(4) |
从(3)式和(4)式可以看出,ρHS2-ρBS2越大,反铁磁性越明显,对于铁磁性相互作用体系,其磁耦合常数就越小。Ruiz等[40]推导出同核多电子体系磁耦合常数与自旋密度的关系(式(5))。张义权等[41]将其推广到异双核多电子体系,得到磁耦合常数与自旋密度的关系,见式(6)。
|
$ \begin{array}{c} \rho _{{\rm{HS}}}^2 - \rho _{{\rm{HS}}}^2 \approx {\Delta _{{\rm{AB}}}} = \\ {\left( {\sqrt {{{\left( {\rho _{{\rm{HS}}}^{\rm{A}}} \right)}^2} - {{\left( {\rho _{{\rm{BS}}}^{\rm{A}}} \right)}^2}} + \sqrt {{{\left( {\rho _{{\rm{HS}}}^{\rm{B}}} \right)}^2} - {{\left( {\rho _{{\rm{BS}}}^{\rm{B}}} \right)}^2}} } \right)^2} \end{array} $ |
(5) |
|
$ \begin{array}{c} \rho _{{\rm{HS}}}^2 - \rho _{{\rm{BS}}}^2 \approx {\Delta _{{\rm{AB}}}} = \\ {\left[ {\sqrt {\left| {{{\left( {\rho _{{\rm{HS}}}^{\rm{A}}} \right)}^2} - {{\left( {\rho _{{\rm{BS}}}^{\rm{A}}} \right)}^2}} \right|} \pm \sqrt {\left| {{{\left( {\rho _{{\rm{HS}}}^{\rm{B}}} \right)}^2} - {{\left( {\rho _{{\rm{BS}}}^{\rm{B}}} \right)}^2}} \right|} } \right]^2} \end{array} $ |
(6) |
式(6)中,当(ρHSA)2-(ρBSA)2和(ρHSB)2-(ρBSB)2符号相同时取正号;当(ρHSA)2-(ρBSA)2和(ρHSB)2-(ρBSB)2的符号相反时取负号。
氧桥联GdNi双核模型配合物磁耦合常数与自旋密度平方差ρHS2-ρBS2的关系如表 5和图 6所示。
下载:
导出CSV
| 键角θ/° | Jcacl/cm-1 | ρHSGd | ρBSGd | ρHSNi | ρBSNi | ρHS2-ρBS2 |
| 85 | -1.62 | 6.969853 | 7.000766 | 1.632451 | -1.624595 | 0.2472 |
| 90 | -0.76 | 6.986042 | 7.007849 | 1.658835 | -1.653578 | 0.1768 |
| 95 | 0.20 | 6.999916 | 7.014368 | 1.677123 | -1.674092 | 0.1220 |
| 100 | 0.96 | 7.011005 | 7.012942 | 1.687259 | -1.683040 | 0.0021 |
| 105 | 1.36 | 7.018669 | 7.019989 | 1.690620 | -1.689782 | 0.0069 |
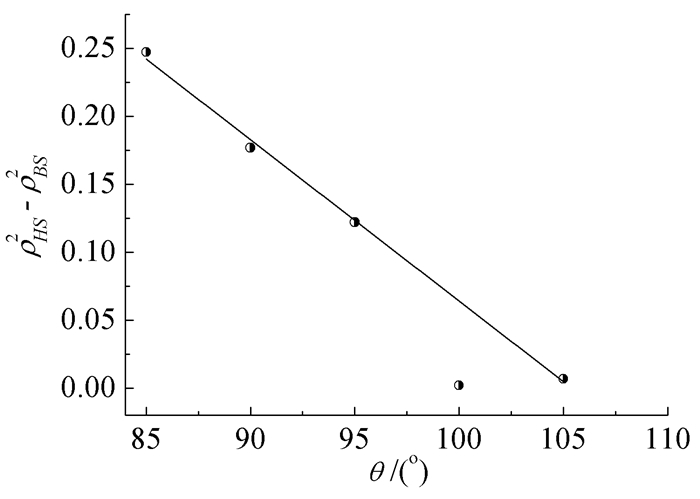
由表 5和图 6可知,在氧桥联GdNi双核模型配合物中,随着Gd-O-Ni键角θ增加,顺磁中心HS态和BS态自旋密度的平方差ρHS2-ρBS2随之减小,配合物顺磁中心间反铁磁性贡献减小,其磁耦合常数Jcalc值随之增大。
本文采用DFT-BS方法研究氧桥联GdNi双核配合物磁耦合作用及机理,结果显示,B3LYP泛函计算的结果与实验值较吻合,能够准确地描述氧桥联GdNi双核配合物的磁学性质。磁轨道分析表明,顺磁中心GdⅢ、NiⅡ与两个桥联原子(O(3),O(4))和醋酸桥氧原子(O(5),O(6))之间存在较强的轨道相互作用。磁轨道主要由顺磁中心NiⅡ的3dz2和3dx2-y2轨道、酚氧桥联配体中氧原子的2pz轨道、醋酸桥中氧原子的2pz轨道和顺磁中心GdⅢ的4fxyz、4fz2x轨道组成。自旋布居分析显示,顺磁中心GdⅢ主要是自旋极化作用,顺磁中心NiⅡ主要是自旋离域作用。随着Gd-O-Ni键角θ的增加,顺磁中心HS态和BS态自旋密度的平方差ρHS2-ρBS2随之减小,反铁磁性相互作用的贡献减小,配合物磁耦合常数增大。
N K Dutt, K Nag. Inorg. Nucl. Chem., 1968, 30(10):2493~2499. http://www.sciencedirect.com/science/article/pii/0022190268802635
Q W Xie, S Q Wu, C M Liu et al. Dalton Transac., 2013, 42(31):11227~11223. doi: 10.1039/c3dt50818h
J P Costes, L Vendier. Eur. J. Inorg. Chem., 2010, (18):2768~2773.
N Ahmed, C Das, S Vaidya et al. Chem. Eur. J., 2014, 20(44):14235~14239. doi: 10.1002/chem.v20.44
A Upadhyay, N Komatireddy, A Ghirri et al. Dalton Transac., 2013, 43(1):259~266. http://europepmc.org/abstract/med/24100508
M Towatari, K Nishi, T Fujinami et al. Inorg. Chem., 2013, 52(10):6160~6178. doi: 10.1021/ic400594u
L Jiang, Y Liu, X Liu et al. Dalton Transac., 2017, 46(37):12558~12573. doi: 10.1039/C7DT02351K
V Vieru, T D Pasatoiu, L Ungur et al. Inorg. Chem., 2016, 55(23):12158~12171. doi: 10.1021/acs.inorgchem.6b01669
M X Yao, Z X Zhu, X Y Lu et al. Dalton Transac., 2016, 45(26):10689~10695. doi: 10.1039/C6DT01606E
T Gupta, M F Beg, G Rajaraman. Inorg. Chem., 2016, 55(21):11201~11215. doi: 10.1021/acs.inorgchem.6b01831
M Maity, M C Majee, S Kundu et al. Inorg. Chem., 2015, 54(20):9715~9726. doi: 10.1021/acs.inorgchem.5b01142
P E Moreno, N F Chilton, F Tuna et al. Inorg. Chem., 2015, 54(12):5930~5941. doi: 10.1021/acs.inorgchem.5b00746
A A Patrascu, S Calancea, M Briganti et al. Chem. Commun., 2017, 53(48):6504~6507. doi: 10.1039/C7CC03236F
W K Dong, J C Ma, Y J Dong et al. Polyhedron, 2016, 115:228~235. doi: 10.1016/j.poly.2016.05.017
L X Zhou, J Q Xu, Y Q Zheng et al. J. Coord. Chem., 2017, 70(19):1~26.
J P Costes, F Dahan, A Dupuis et al. Inorg. Chem., 1997, 36(16):3429~3433. doi: 10.1021/ic970264v
O Roubeau, G Lorusso, S J Teat et al. Dalton Transac., 2014, 43(30):11502-11509. doi: 10.1039/C4DT00697F
E Colacio, J Ruiz, A J Mota et al. Inorg. Chem., 2012, 51(10):5857~5868. doi: 10.1021/ic3004596
Q Y Chen, Q H Luo, L M Zheng et al. Inorg. Chem., 2002, 41(3):605~609. doi: 10.1021/ic010270y
T D Pasatoiu, J P Sutter, A M Madalan et al. Inorg. Chem., 2011, 50(13):5890~5898. doi: 10.1021/ic2004276
S K Singh, N K Tibrewal, G Rajaraman. Dalton Transac., 2011, 40(41):10897~10906. doi: 10.1039/c1dt10600g
J Paulovič, F Cimpoesu, M Ferbinteanu et al. J. Am. Chem. Soc., 2004, 126(10):3321~3331. doi: 10.1021/ja030628k
X L Li, F Y Min, C Wang et al. Inorg. Chem., 2015, 54(9):4337~4344. doi: 10.1021/acs.inorgchem.5b00019
E Cremades, S Gómez-coca, D Aravena et al. J. Am. Chem. Soc., 2012, 134(25):10532~10542. doi: 10.1021/ja302851n
J P Costes, F Dahan, A Dupuis et al. Inorg. Chem. 1997, 36:4284~4286. doi: 10.1021/ic970720f
S K Singh, T Rajeshkumar, V Chandrasekhar et al. Polyhedron, 2013, 66(1):81~86. http://www.sciencedirect.com/science/article/pii/S0277538713001381
黎健.化学学报, 2000, 58(12):1529~1533. doi: 10.3321/j.issn:0567-7351.2000.12.008
F Neese. WIREs Comput. Mol. Sci., 2012, 2(1):73~78. doi: 10.1002/wcms.81
F Neese. J. Phys. Chem. Solids, 2004, 65(4):781~785. doi: 10.1016/j.jpcs.2003.11.015
罗树常, 刘翔宇, 张竹霞等.分子科学学报, 2017, 33(2):120~126. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=FZKB201702006&dbname=CJFD&dbcode=CJFQ
曹景阳, 孙小媛, 罗树常等.广东化工, 2017, 44(7):130~132. doi: 10.3969/j.issn.1007-1865.2017.07.056
W Humphrey, A Dalke, K Schulten. J. Molec. Graphics, 1996, 14(1):33~38. doi: 10.1016/0263-7855(96)00018-5
D A Pantazis, F Neese. J. Chem. Theory Comput., 2009, 5(9):2229~2238. doi: 10.1021/ct900090f
胡宗超, 卫海燕, 王凡等.化学学报, 2004, 62(20):1973~1980. doi: 10.3321/j.issn:0567-7351.2004.20.001
J Y Bian, Y F Chang, J P Zhang. J. Phys. Chem. A, 2008, 112(14):3186~3191. doi: 10.1021/jp711121z
阎峰, 陈志达.北京大学学报(自然科学版), 2000, 36(6):873~880. doi: 10.3321/j.issn:0479-8023.2000.06.021
F Yan, Z D Chen. J. Phys. Chem. A, 2000, 104(26):6295~6300. doi: 10.1021/jp994093m
F Illas, R Caballol, O Castell et al. J. Phys. Chem. A, 1997, 101(42):7860~7866. doi: 10.1021/jp9711757
E Ruiz, J Cano, S Alvarez et al. J. Comp. Chem., 1999, 20(13):1391~1400. doi: 10.1002/(ISSN)1096-987X
T Cauchy, E Ruiz, S Alvarez. J. Am. Chem. Soc., 2006, 128(49):15722~15727. doi: 10.1021/ja0641498
Y Q Zhang, C L Luo. Eur. J. Inorg. Chem., 2008(13):2199~2206.

图 1 配合物[Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]分子结构
Figure 1 Molecular structure of complex [Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]

图 2 配合物[Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]十重态的单占据磁轨道(SOMOs)
Figure 2 Singly occupied molecular orbitals (SOMOs) for title complex in decuplet state

图 3 配合物[Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2]六重态的局域分子磁轨道
Figure 3 Local magnetic orbital for title complex in sextet state

图 4 配合物[Ni(3-MeOsaltn)(MeOH)(ac)Gd(hfac)2] HS和BS态的自旋密度图
Figure 4 Spin density maps for title complex in HS and BS state

图 5 氧桥联GdNi双核配合物磁耦合常数Jcalc值与键角的关系
Figure 5 The relationship between magnetic coupling constants 2Jcalc and bond angles (θ) for oxygen bridged dinuclear GdⅢNiⅡ complexes

图 6 配合物磁耦合常数(Jcacl)与自旋密度的平方差的关系
Figure 6 The relationships between magnetic coupling constants 2Jcalc and ρHS2-ρBS2 for title complexes
表 1 不同泛函计算HS、BS态能量(EHS、EBS)、自旋平方算符的期望值(S2)及磁耦合常数(Jcacl)
Table 1. Total energies (EHS, EBS), expectation value of the total spin angular momentum squared (S2) for sextet states (BS) and decuplet (HS) states, and the magnetic coupling constant (Jcalc) values for title complex obtained by several DFT methods with TZVP (Gd/SARC2-DKH-QZVP) basis set
| 泛函 | EHS/eV | EBS/eV | SHS2 | SBS2 | J/cm-1 |
| LDA | -438845.7838 | -438845.7709 | 24.8425 | 10.8131 | 7.39 |
| VWN3 | -439129.5037 | -439129.4907 | 24.8394 | 10.8047 | 7.47 |
| VWN5 | -438845.7838 | -438845.7709 | 24.8425 | 10.8131 | 7.39 |
| BLYP | -439833.5984 | -439833.5906 | 24.8133 | 10.7845 | 4.48 |
| XLYP | -439929.0020 | -439928.9946 | 24.8111 | 10.7820 | 4.28 |
| BP86 | -439872.0800 | -439871.9861 | 25.1168 | 11.1705 | 54.30 |
| TPSS | -439808.5077 | -439808.5058 | 24.7635 | 10.7632 | 1.12 |
| TPSSh | -439824.9913 | -439824.9886 | 24.7663 | 10.7654 | 1.59 |
| TPSS0 | -439808.5077 | -439808.5058 | 24.7635 | 10.7632 | 1.12 |
| M06 | -439793.4064 | -439793.4048 | 24.7687 | 10.7684 | 0.94 |
| M06-L | -439815.5809 | -439815.5775 | 24.7849 | 10.7807 | 1.99 |
| M06-2X | -439830.6805 | -439830.6791 | 24.7666 | 10.7664 | 0.79 |
| B2PLYP | -439634.3555 | -439634.3545 | 24.7627 | 10.7626 | 0.57 |
| mPW2PLYP | -439645.5448 | -439645.5438 | 24.7627 | 10.7625 | 0.55 |
| PWPB95 | -439704.1563 | -439704.1552 | 24.7622 | 10.7621 | 0.62 |
| B3LYP | -439786.7372 | -439786.7352 | 24.7638 | 10.7634 | 1.18 |
| B3P86 | -439825.1466 | -439825.1444 | 24.7648 | 10.7643 | 1.29 |
| X3LYP | -439762.2324 | -439762.2304 | 24.7636 | 10.7632 | 1.13 |
| O3LYP | -439729.1774 | -439729.1745 | 24.7731 | 10.7717 | 1.68 |
| PBE0 | -439722.6541 | -439722.6521 | 24.7641 | 10.7638 | 1.16 |
| mPW1LYP | -439810.6793 | -439810.6774 | 24.7630 | 10.7626 | 1.04 |
 下载: 导出CSV
下载: 导出CSV
表 2 B3LYP下不同基组计算HS、BS态能量(EHS、EBS)及磁耦合常数(Jcacl)
Table 2. Total energies (EHS, EBS), expectation value of the total spin angular momentum squared (S2) for sextet states (BS) and decuplet (HS) states, and the magnetic coupling constant (Jcalc) values for title complex obtained by several basis sets with B3LYP method
| 基组 | EHS/eV | EBS/eV | SHS2 | SBS2 | Jcalc/cm-1 |
| Gd(SARC2-DKH-QZVP) 其他原子(QZVP) |
-439828.5262 | -439828.5242 | 24.7636 | 10.7631 | 1.15 |
| Gd(SARC2-DKH-QZVP) 其他原子(TZVP) |
-439786.7372 | -439786.7352 | 24.7638 | 10.7634 | 1.18 |
| Gd(SARC-DKH-TZVPP) 其他原子(TZVP) |
-439753.0795 | -439753.0774 | 24.7638 | 10.7633 | 1.18 |
| Gd(SARC-DKH-TZVP) 其他原子(TZVP) |
-439753.0795 | -439753.0774 | 24.7638 | 10.7633 | 1.18 |
| Gd(SARC-DKH-TZVP) 其他原子(TZV) |
-439727.9009 | -439727.8989 | 24.7641 | 10.7636 | 1.16 |
下载: 导出CSV
表 3 B3LYP/TZVP(Gd为SARC2-DKH-QZVP)水平下不同配合物计算HS、BS态能量(EHS、EBS)及磁耦合常数(Jcacl)
Table 3. Total energies (EHS, EBS), expectation value of the total spin angular momentum squared (S2) for sextet states (BS) and decuplet (HS) states, and the magnetic coupling constant (Jcalc) values for complexes obtained at B3LYP/TZVP (Gd/SARC2-DKH-QZVP) level
| Complexes | EHS/eV | EBS/eV | SHS2 | SBS2 | Jcacl/cm-1 | Jexp/cm-1 |
| [(C21H24N2O4)Ni(H2O)2Gd(NO3)3] | -408402.5386 | -408402.5362 | 24.7607 | 10.7604 | 1.34 | 1.8[35] |
| [Ni(CH3CN)2(valpn)Gd(NO3)3]3·CH3CN | -409316.1196 | -409316.1171 | 24.7620 | 10.7616 | 1.46 | 1.15[20] |
| [(NiL)Gd(hfac)2(EtOH)]·1.5EtOH | -441419.2514 | -441419.2508 | 24.7619 | 10.7615 | 0.33 | 0.34[2] |
| [Ni(HL)(H2O)(tfa)Gd(hfac)2] | -448879.4516 | -448879.4493 | 24.7611 | 10.7607 | 1.32 | 1.83[7] |
| [Ni(valpan)(MeOH)(ac)Ln(hfac)2] | -439727.9010 | -439727.8989 | 24.7641 | 10.7636 | 1.16 | 1.10[6] |
| [Ni(μ-L)(μ-Ac)Gd(NO3)2] | -408668.1892 | -408668.1882 | 24.7599 | 10.7593 | 0.55 | 0.69[18] |
下载: 导出CSV
表 4 配合物BS态的自旋电子在原子轨道上的分布
Table 4. spin electron distribution on atomic orbitals for complex's BS state
| 原子 | 原子轨道 | |||
| s | p | d | f | |
| Gd(1) | 0.0074 | 0.0125 | 0.0508 | 6.9469 |
| Ni(2) | 0.0024 | 0.0167 | -1.6666 | |
| O(3) | -0.0055 | -0.0364 | ||
| O(4) | -0.0058 | -0.0360 | ||
| O(5) | -0.0003 | -0.0016 | ||
| O(6) | -0.0042 | -0.0347 | ||
| O(7) | -0.0038 | -0.1102 | ||
| N(8) | -0.0173 | -0.0426 | ||
| N(9) | -0.0169 | -0.0435 | ||
| O(10) | -0.0004 | -0.0025 | ||
| O(11) | -0.0004 | -0.0023 | ||
| O(12) | -0.0002 | -0.0014 | ||
| O(13) | -0.0003 | -0.0030 | ||
| O(14) | -0.0003 | -0.0031 | ||
| O(15) | -0.0002 | -0.0017 | ||
下载: 导出CSV
表 5 配合物磁耦合常数(Jcacl)与自旋密度的平方差的关系
Table 5. The relationships between magnetic coupling constants 2Jcalc and ρHS2-ρBS2for title complexes
| 键角θ/° | Jcacl/cm-1 | ρHSGd | ρBSGd | ρHSNi | ρBSNi | ρHS2-ρBS2 |
| 85 | -1.62 | 6.969853 | 7.000766 | 1.632451 | -1.624595 | 0.2472 |
| 90 | -0.76 | 6.986042 | 7.007849 | 1.658835 | -1.653578 | 0.1768 |
| 95 | 0.20 | 6.999916 | 7.014368 | 1.677123 | -1.674092 | 0.1220 |
| 100 | 0.96 | 7.011005 | 7.012942 | 1.687259 | -1.683040 | 0.0021 |
| 105 | 1.36 | 7.018669 | 7.019989 | 1.690620 | -1.689782 | 0.0069 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们