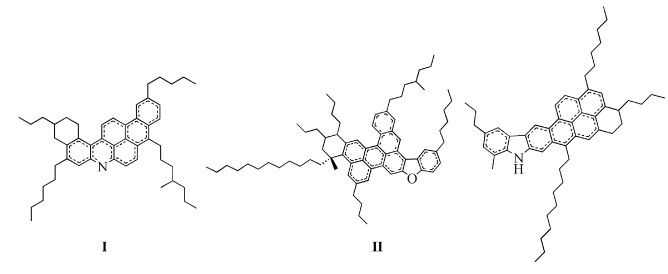
图式 1.
3种沥青质分子结构模型
Scheme 1.
Three molecular structure models of asphaltene

石油作为当今世界上最重要的地质勘探对象之一,是储存于地壳上层部分的深褐色粘稠液体,含有烷烃、环烷烃和芳香烃等组分。虽然全球石油资源的探明储量在近几年均呈现微增长,但增长的部分大多是页岩油和重质油等非常规石油资源,而这类石油由于开采难度大且开采成本较高,利用率低。在不久的未来,石油资源将越发稀缺,非常规石油资源将会在能源市场上占有重要一席,并会具有更大的利用价值,所以对其研究尤为重要。
重质油作为非常规石油资源的重要组成部分,是指用常规原油开采技术难于开采的具有较大粘度和密度的原油。重质油是含有氧、硫、氮和金属元素等杂原子的以芳香分为中心、周围附以脂肪环及脂肪支链的结构极其复杂的化合物[1],其密度在931~998 kg/m3之间[2]。由于地理环境与气候的不同,导致不同地域的重质原油组分略有差异,按分子量大小可分为饱和分、芳香分、胶质和沥青质,这四种组分按极性强弱排序为沥青质>胶质>芳香分>饱和分,其中极性最强且分子量最大的组分是沥青质,它的组成与结构在很大程度上决定了重质原油的相分离、流变性、原油乳状液稳定性、浸润性[3~10]等性质。
在石油的生产、运输和加工过程中,沥青质容易发生聚沉生焦,严重影响重质原油的加工利用率,因而需要深化对沥青质大分子的认识。
由于重质油的组分结构极其复杂,鉴于现今检测技术的局限性,对重质油中各组分的结构和性质认识也只是预测性的[11~19]。
(1) 分子间相互作用。近年来,国外科学家通过实验与检测手段对沥青质宏观结构进行了研究并提出了沥青质大分子的聚合薄片模型。这种模型是以芳香片段为中心、烷基侧链连接的大分子结构,由于芳香薄片的π-π作用聚集在一起。如图式 1所示,Groenzin等[19]提出了一些沥青质结构模型,在这些模型中有一些杂原子(例如O、S和N等),而这些杂原子也被证明在其他沥青质模型中普遍存在的。本文通过参考图式 1的结构模型,选取其中的分子片段,研究其间相互作用,旨在考查沥青质大分子之间除芳香组分π-π纵向作用之外的相互作用。由于沥青质结构复杂,其中具有强电负性的杂原子(如O、S、N等)可与其他沥青质大分子的芳香环发生相互作用,也可与脂肪饱和环和烷基侧链发生相互作用,若都讨论的话种类繁多且计算量巨大,所以在此暂且忽略饱和环与烷基侧链的影响,通过讨论杂环之间的相互作用试图得到沥青质间相互作用的有用信息。
本文构建了氧芴与氧芴、氧芴与吖啶、氧芴与咔唑这三组空间构型,在M062X/6-31G(d)水平上对各组不同构型进行了结构优化和频率振动分析,由结构分析、前线轨道能量及能级差、分子间相互作用能得到各组中最稳定的二元相互作用构型。
(2) 溶剂化效应。原油中的沥青质容易发生聚沉导致石油的流动性降低、引发油藏的坏死和输油管线的堵塞等严重问题。因此,进行重质油沥青质的溶解性研究对沥青质聚沉的预防和聚沉抑制剂的开发具有重要指导意义。实验表明,一些溶剂能够抑制沥青质的聚沉甚至解除沥青质聚合物[20~22]。但国内的相关研究大多是以储集层的孔隙结构为重点[23,24],关于沥青质聚沉微观机理的研究却鲜有报道。国外对重质油沥青质在不同种溶剂中的聚合物解聚的相关研究主要有3种方法:距离法[25]、溶解度参数法[26]和能量法[27]。
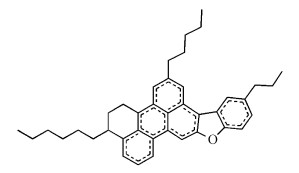
本文将从溶剂化自由能的角度和静电、非静电作用分析对不同溶剂中沥青质的溶解性进行定量研究。选取上述图式 1所示的两个沥青质结构模型Ⅰ和Ⅱ作为本文的模型分子。这类沥青质模型是以芳香组分为核心,与饱和脂肪环和烷基侧链相连,并且带有O、S、N等杂原子的大分子构型。模型分子Ⅰ为带有N原子与吖啶(二苯并吡啶)分子的沥青质大分子。模型分子Ⅱ为带有O原子与氧芴(二苯并呋喃)的大分子,但由于此分子结构复杂且分子量巨大,进行模型优化与溶剂化计算时过于耗时,因此对该分子进行适当简化,如图式 2中分子Ⅲ所示,简化后的分子同样由芳香分、脂肪环与烷基侧链连接构成,其间含有O原子与氧芴分子,即仍保有原分子的构型特点。
运用密度泛函理论(DFT)中的B3LYP方法,选用SMD溶剂化模型计算沥青质模型分子Ⅰ和经简化后的模型分子Ⅲ在13种溶剂中的溶剂化自由能。针对重质油中沥青质容易产生聚沉这一问题,试图找到预防沥青质聚沉和研究聚沉抑制剂性质的相关信息。
运用DFT中的M062X[28]方法,结合6-31G(d)基组,对氧芴、吖啶、咔唑单体以及混合二元体系可能存在的几何构型进行建模,并进行结构优化,寻找可能存在的稳定构型。并在6-31G(d)基组水平上做振动频率分析,确定优化后的构型无虚频且为势能面上的极小值。采用自然键轨道(NBO)[29]方法分析了电荷分布变化,并对混合体系构型进行Mulliken重叠布居分析。
在6-31G(d)和6-311++G(2d,2p)这2种基组水平上,通过单体和混合体系之间的能量差计算了分子间的相互作用能(采用M062X方法计算分子间相互作用时不需进行基组叠加误差BSSE校正[30],其计算结果较进行BSSE校正后的结果更准确)。对于A+B混合体系的相互作用能为:
|
$ \Delta E = {E_{{\rm{A + B}}}}-{E_{\rm{A}}}-{E_{\rm{B}}} $ |
式中,EA+B为混合体系的总能量;EA为单体A的能量;EB为单体B的能量。
对于静态模拟的溶剂化效应主要关注两个方面:一个是极性溶剂的偶极矩与溶质分子偶极矩之间的静电相互作用,这种属于远程作用;另一是溶剂分子与溶质分子反应中心有键的作用,包括配位键和氢键等,这种作用属于短程作用。色散力在溶剂与溶质之间也有重要的远程作用,尤其对于非极性溶剂而言,但是利用化学模拟技术表示色散力是很困难的,所以暂不予考虑[31]。
虚拟溶剂模型通常把溶剂效应看成是溶质分子分布在具有均一性质的连续介质当中,也称为反应场。短程作用与远程作用的连续介质模型结合起来的方法综合考虑了溶剂的短程作用和远程作用[32]。不同于Gaussian03,Gaussian09引入了SMD模型,SMD溶剂模型包含了静电作用与非静电作用两部分,静电部分是使用特定参数下做IEFPCM (PCM using the Intergral Equation Formalism model)计算,非静电部分是根据原子类型和接触面积近似得到的,目的在于得到准确的溶剂化自由能。
本部分使用DFT中的B3LYP[33,34]方法,结合6-31G(d)基组,首先对沥青质模型分子Ⅰ和经简化的模型分子Ⅲ进行构型建模、结构优化和频率振动分析,再选用SMD溶剂化模型计算模型分子Ⅰ和模型分子Ⅲ在13种溶剂(含乙腈、苯、氯苯、三氯甲烷、环己烷、二氯乙烷、二氯甲烷、二乙醚、乙醇、正庚烷、甲醇、四氢呋喃和甲苯)中的静电溶剂化自由能(ΔGelec)、非静电溶剂化自由能(ΔGnonelec)、总溶剂化自由能(ΔGsolv)来研究其溶剂化性质。
以上所有的计算工作由Gaussian 09程序完成。
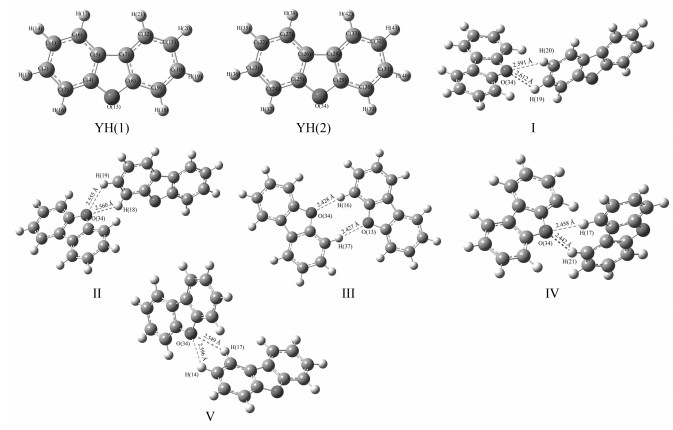
经优化与振动频率分析确定的稳定构型有11种,分为3组。其中第一组构型为氧芴分子与氧芴分子形成的混合体系,序号为Ⅰ至Ⅴ;第二组构型为氧芴分子与吖啶分子形成的混合体系,序号为Ⅵ至Ⅹ;第三组构型为氧芴分子与咔唑分子形成的混合体系,序号为Ⅺ。
氧芴分子单体与混合体系的优化构型如图 1所示,5种混合构型形成了不同的氢键,均为氧芴中的O原子与对方分子苯环上的H原子间存在相互作用;在Ⅰ、Ⅱ、Ⅳ、Ⅴ这4个构型中,氧芴的O与对方分子的相邻2个H同时产生氢键,这4种构型中,只有一个氧芴中的O原子参与氢键的形成。而在构型Ⅲ中,两个分子的O分别与对方分子的H产生氢键,即两边氧芴分子的O原子均参与了氢键的形成;氧芴单体分子是平面分子,由图可见,构型Ⅲ中两个分子的空间排布接近于同一平面,而其余4种构型的两个分子平面明显存在很大夹角;这组构型中,分子间距大小顺序依次为Ⅲ(2.427Å)<Ⅳ(2.442Å)<Ⅴ(2.549Å)<Ⅱ(2.555Å)<Ⅰ(2.591Å),可推断5种构型中分子间相互作用最强的是构型Ⅲ。
氧芴、吖啶分子单体与混合体系的全优化构型如图 2所示,5种稳定构型为Ⅵ~Ⅹ,不同的构型有不同的氢键。在Ⅵ、Ⅶ、Ⅸ、Ⅹ中,吖啶分子的N与氧芴分子中相邻的2个H存在相互作用,这4种构型中,只有吖啶分子的N原子参与到氢键的形成。构型Ⅷ中,分子间氢键既存在于吖啶分子的N与氧芴分子的H之间,也存在于吖啶分子的H与氧芴分子的O之间,即两边杂环分子的杂原子均参与了氢键的形成;氧芴分子与吖啶分子均是平面型分子,由图可见,构型Ⅷ中氧芴与吖啶由于氢键作用,呈现出的空间构型接近于同一平面;这组构型中,分子间距大小顺序依次为Ⅷ(2.350Å)<Ⅸ(2.507Å)<Ⅹ(2.580Å)<Ⅵ(2.587Å)<Ⅶ(2.601Å)。可推测这一组5种构型中分子间相互作用最强的是构型Ⅷ。
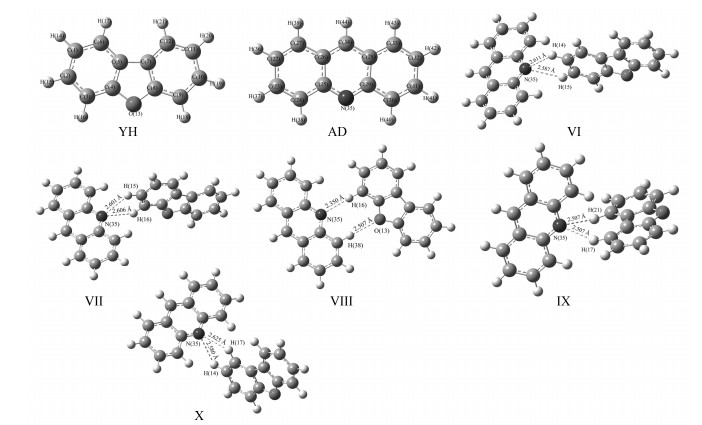
图 3是氧芴、咔唑分子单体与稳定混合体系的优化构型,可见氧芴分子的O和与咔唑分子N原子相连的H存在氢键,同时咔唑分子上的N与氧芴分子苯环上的H存在氢键。
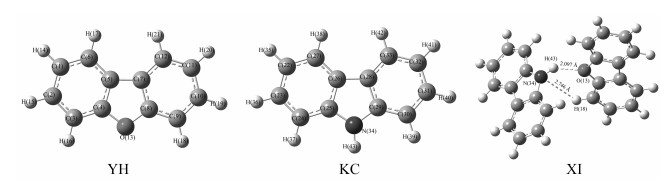
表 1列出了氧芴和混合体系的原子NBO电荷,表 2列出了氧芴、吖啶、咔唑及混合体系的原子NBO电荷。电荷变化较多的原子是体系中参与相互作用的原子,且11种优化构型中均有不同程度的原子间电荷转移。例如,相对于氧芴单体分子,混合体系Ⅰ中的O(34)减少了0.014e,H(19)增加了0.009e,H(20)增加了0.010e;体系Ⅷ中,N(35)减少了0.020e,H(16)增加了0.022e,O(13)减少了0.009e,H(38)增加了0.009e,吖啶分子的净电荷相对增加了0.008e;体系Ⅺ中,O(13)减少了0.035e,H(18)增加了0.011e,N(34)减少了0.009e,H(43)增加了0.015e。体系中其他未参与相互作用的原子电荷变化都不大,结果表明大部分的电荷转移是因为分子间存在一定相互作用。
 下载:
导出CSV
下载:
导出CSV
| Atom | Isolated molecule | Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ |
| C(1) | -0.259 | -0.259 | -0.259 | -0.258 | -0.257 | -0.267 |
| C(2) | -0.231 | -0.231 | -0.231 | -0.230 | -0.231 | -0.231 |
| C(3) | -0.281 | -0.281 | -0.281 | -0.280 | -0.282 | -0.282 |
| C(4) | 0.333 | 0.332 | 0.332 | 0.331 | 0.331 | 0.331 |
| C(5) | -0.112 | -0.112 | -0.112 | -0.113 | -0.118 | -0.113 |
| C(6) | -0.210 | -0.210 | -0.210 | -0.211 | -0.215 | -0.215 |
| C(7) | -0.112 | -0.114 | -0.114 | -0.110 | -0.119 | -0.112 |
| C(8) | 0.333 | 0.331 | 0.331 | 0.332 | 0.331 | 0.331 |
| C(9) | -0.281 | -0.281 | -0.288 | -0.281 | -0.282 | -0.281 |
| C(10) | -0.231 | -0.237 | -0.236 | -0.231 | -0.231 | -0.231 |
| C(11) | -0.259 | -0.267 | -0.259 | -0.258 | -0.258 | -0.259 |
| C(12) | -0.210 | -0.211 | -0.211 | -0.210 | -0.216 | -0.209 |
| O(13) | -0.474 | -0.475 | -0.474 | -0.485 | -0.475 | -0.475 |
| H(14) | 0.246 | 0.246 | 0.246 | 0.246 | 0.246 | 0.255 |
| H(15) | 0.247 | 0.247 | 0.247 | 0.245 | 0.246 | 0.247 |
| H(16) | 0.258 | 0.258 | 0.258 | 0.272 | 0.257 | 0.257 |
| H(17) | 0.246 | 0.246 | 0.246 | 0.246 | 0.258 | 0.254 |
| H(18) | 0.258 | 0.258 | 0.265 | 0.255 | 0.257 | 0.257 |
| H(19) | 0.247 | 0.256 | 0.254 | 0.247 | 0.246 | 0.246 |
| H(20) | 0.246 | 0.256 | 0.246 | 0.246 | 0.246 | 0.246 |
| H(21) | 0.246 | 0.246 | 0.246 | 0.247 | 0.258 | 0.248 |
| C(22) | -0.259 | -0.257 | -0.257 | -0.258 | -0.256 | -0.257 |
| C(23) | -0.231 | -0.228 | -0.230 | -0.230 | -0.226 | -0.228 |
| C(24) | -0.281 | -0.279 | -0.280 | -0.280 | -0.275 | -0.280 |
| C(25) | 0.333 | 0.330 | 0.332 | 0.331 | 0.328 | 0.329 |
| C(26) | -0.112 | -0.112 | -0.111 | -0.113 | -0.111 | -0.112 |
| C(27) | -0.210 | -0.211 | -0.210 | -0.211 | -0.211 | -0.211 |
| C(28) | -0.112 | -0.111 | -0.112 | -0.110 | -0.109 | -0.111 |
| C(29) | 0.333 | 0.331 | 0.330 | 0.332 | 0.330 | 0.331 |
| C(30) | -0.281 | -0.281 | -0.280 | -0.281 | -0.281 | -0.281 |
| C(31) | -0.231 | -0.230 | -0.228 | -0.231 | -0.230 | -0.230 |
| C(32) | -0.259 | -0.258 | -0.257 | -0.258 | -0.257 | -0.257 |
| C(33) | -0.210 | -0.210 | -0.210 | -0.210 | -0.210 | -0.210 |
| O(34) | -0.474 | -0.488 | -0.490 | -0.485 | -0.493 | -0.489 |
| H(35) | 0.246 | 0.246 | 0.247 | 0.246 | 0.247 | 0.247 |
| H(36) | 0.247 | 0.248 | 0.248 | 0.245 | 0.250 | 0.249 |
| H(37) | 0.258 | 0.266 | 0.259 | 0.272 | 0.264 | 0.265 |
| H(38) | 0.246 | 0.246 | 0.247 | 0.246 | 0.247 | 0.247 |
| H(39) | 0.258 | 0.258 | 0.264 | 0.255 | 0.259 | 0.259 |
| H(40) | 0.247 | 0.248 | 0.248 | 0.247 | 0.248 | 0.248 |
| H(41) | 0.246 | 0.247 | 0.247 | 0.246 | 0.247 | 0.247 |
| H(42) | 0.246 | 0.247 | 0.247 | 0.247 | 0.247 | 0.247 |
下载:
导出CSV
| Atom | Isolated | Ⅵ | Ⅶ | Ⅷ | Ⅸ | Ⅹ | Atom | Isolated | Ⅺ |
| molecule | molecule | ||||||||
| C(1) | -0.259 | -0.263 | -0.260 | -0.258 | -0.260 | -0.263 | C(1) | -0.259 | -0.255 |
| C(2) | -0.231 | -0.235 | -0.234 | -0.233 | -0.232 | -0.231 | C(2) | -0.231 | -0.228 |
| C(3) | -0.281 | -0.283 | -0.286 | -0.281 | -0.284 | -0.283 | C(3) | -0.281 | -0.281 |
| C(4) | 0.333 | 0.330 | 0.330 | 0.331 | 0.330 | 0.329 | C(4) | 0.333 | 0.330 |
| C(5) | -0.112 | -0.115 | -0.116 | -0.114 | -0.117 | -0.115 | C(5) | -0.112 | -0.108 |
| C(6) | -0.210 | -0.212 | -0.212 | -0.214 | -0.214 | -0.216 | C(6) | -0.210 | -0.208 |
| C(7) | -0.112 | -0.111 | -0.112 | -0.110 | -0.117 | -0.111 | C(7) | -0.112 | -0.110 |
| C(8) | 0.333 | 0.331 | 0.332 | 0.333 | 0.330 | 0.331 | C(8) | 0.333 | 0.325 |
| C(9) | -0.281 | -0.281 | -0.281 | -0.282 | -0.284 | -0.282 | C(9) | -0.281 | -0.278 |
| C(10) | -0.231 | -0.233 | -0.233 | -0.232 | -0.232 | -0.233 | C(10) | -0.231 | -0.225 |
| C(11) | -0.259 | -0.260 | -0.260 | -0.259 | -0.260 | -0.260 | C(11) | -0.259 | -0.254 |
| C(12) | -0.210 | -0.211 | -0.211 | -0.210 | -0.214 | -0.210 | C(12) | -0.210 | -0.210 |
| O(13) | -0.474 | -0.476 | -0.475 | -0.483 | -0.477 | -0.476 | O(13) | -0.474 | -0.509 |
| H(14) | 0.246 | 0.258 | 0.244 | 0.244 | 0.245 | 0.259 | H(14) | 0.246 | 0.248 |
| H(15) | 0.247 | 0.261 | 0.259 | 0.243 | 0.245 | 0.245 | H(15) | 0.247 | 0.250 |
| H(16) | 0.258 | 0.256 | 0.271 | 0.280 | 0.256 | 0.256 | H(16) | 0.258 | 0.259 |
| H(17) | 0.246 | 0.244 | 0.244 | 0.245 | 0.263 | 0.260 | H(17) | 0.246 | 0.249 |
| H(18) | 0.258 | 0.257 | 0.257 | 0.255 | 0.256 | 0.257 | H(18) | 0.258 | 0.269 |
| H(19) | 0.247 | 0.246 | 0.246 | 0.246 | 0.245 | 0.246 | H(19) | 0.247 | 0.251 |
| H(20) | 0.246 | 0.245 | 0.245 | 0.245 | 0.245 | 0.245 | H(20) | 0.246 | 0.248 |
| H(21) | 0.246 | 0.246 | 0.245 | 0.246 | 0.263 | 0.248 | H(21) | 0.246 | 0.248 |
| C(22) | -0.240 | -0.239 | -0.239 | -0.238 | -0.237 | -0.239 | C(22) | -0.270 | -0.271 |
| C(23) | -0.235 | -0.231 | -0.231 | -0.232 | -0.227 | -0.231 | C(23) | -0.231 | -0.232 |
| C(24) | -0.221 | -0.223 | -0.223 | -0.222 | -0.219 | -0.224 | C(24) | -0.279 | -0.281 |
| C(25) | 0.191 | 0.195 | 0.195 | 0.196 | 0.197 | 0.195 | C(25) | 0.181 | 0.173 |
| C(26) | -0.104 | -0.104 | -0.104 | -0.103 | -0.103 | -0.104 | C(26) | -0.093 | -0.095 |
| C(27) | -0.212 | -0.211 | -0.211 | -0.213 | -0.212 | -0.211 | C(27) | -0.206 | -0.208 |
| C(28) | -0.104 | -0.104 | -0.104 | -0.104 | -0.105 | -0.104 | C(28) | -0.093 | -0.094 |
| C(29) | 0.191 | 0.195 | 0.195 | 0.195 | 0.195 | 0.195 | C(29) | 0.181 | 0.180 |
| C(30) | -0.221 | -0.223 | -0.223 | -0.223 | -0.225 | -0.224 | C(30) | -0.279 | -0.278 |
| C(31) | -0.235 | -0.231 | -0.231 | -0.232 | -0.230 | -0.231 | C(31) | -0.231 | -0.233 |
| C(32) | -0.240 | -0.239 | -0.239 | -0.240 | -0.240 | -0.239 | C(32) | -0.270 | -0.271 |
| C(33) | -0.212 | -0.211 | -0.211 | -0.210 | -0.210 | -0.211 | C(33) | -0.206 | -0.207 |
| C(34) | -0.144 | -0.140 | -0.140 | -0.140 | -0.138 | -0.140 | N(34) | -0.585 | -0.594 |
| N(35) | -0.438 | -0.465 | -0.464 | -0.458 | -0.471 | -0.466 | H(35) | 0.243 | 0.242 |
| H(36) | 0.246 | 0.247 | 0.247 | 0.246 | 0.247 | 0.247 | H(36) | 0.243 | 0.243 |
| H(37) | 0.247 | 0.248 | 0.249 | 0.247 | 0.250 | 0.249 | H(37) | 0.242 | 0.250 |
| H(38) | 0.256 | 0.258 | 0.259 | 0.265 | 0.258 | 0.258 | H(38) | 0.243 | 0.242 |
| H(39) | 0.242 | 0.243 | 0.243 | 0.242 | 0.243 | 0.243 | H(39) | 0.242 | 0.243 |
| H(40) | 0.256 | 0.259 | 0.258 | 0.252 | 0.256 | 0.259 | H(40) | 0.243 | 0.242 |
| H(41) | 0.247 | 0.249 | 0.248 | 0.247 | 0.249 | 0.249 | H(41) | 0.243 | 0.242 |
| H(42) | 0.246 | 0.247 | 0.247 | 0.247 | 0.248 | 0.247 | H(42) | 0.243 | 0.242 |
| H(43) | 0.242 | 0.243 | 0.243 | 0.243 | 0.243 | 0.243 | H(43) | 0.439 | 0.454 |
| H(44) | 0.242 | 0.243 | 0.243 | 0.243 | 0.244 | 0.243 |
第一组氧芴+氧芴组成的构型中,因为空间结构与所形成的氢键均不同,导致电荷在分子间的转移程度不同。在Ⅰ、Ⅱ、Ⅳ、Ⅴ中,氧芴分子(原子序号:22~42)的净电荷分别增加0.002e,0.004e,0.008e及0.003e;Ⅲ的净电荷不变。由于是同种分子组成的二元体系,分子之间的电荷转移并不明显,净电荷的变化量并不大,但电荷的转移大多朝向同一个分子;类似地,第二组构型Ⅵ、Ⅶ、Ⅷ、Ⅸ、Ⅹ中,吖啶分子的净电荷分别增加0.006e,0.007e,0.008e,0.013e和0.004e。可见,这组构型的分子间电荷转移较明显,且电荷都是由吖啶分子向氧芴分子转移;第三组构型Ⅺ中,咔唑分子的净电荷减少了0.011e,电荷由氧芴分子向咔唑分子转移。
总之,电荷不仅在特定的原子间不同程度地转移,同时在体系中的分子间发生转移,说明体系中的电子云发生了一定的变化。
布居是指电子在各原子轨道上的分布,分析这种布居,对于了解分子中原子的成键情况有帮助。Mulliken重叠布居表示电荷在各组成原子之间的分布情况[35~37]。其大小可以说明原子间结合力度大小,其值越大,结合力越大。
表 3、表 4给出了在M062X/6-31G(d)水平上计算得到的各个优化构型中部分化学键的重叠布居数。分析数据可以发现,体系中的C—C、C—O、C—N键上的布居数较大,说明这些共价键有较强的相互作用。而体系中形成的O…H与N…H等氢键的重叠布居数较小,表明形成氢键的原子之间的相互作用相对较弱。混合体系Ⅰ中O…H的Mulliken布居数为0.007,0.007;Ⅱ中O…H的Mulliken布居数为0.007,0.008;Ⅲ中O…H的Mulliken布居数为0.017,0.017;Ⅳ中O…H的Mulliken布居数为0.014,0.014;Ⅴ中O…H的Mulliken布居数为0.008,0.008;Ⅵ中N…H的Mulliken布居数为0.012,0.013;Ⅶ中N…H的Mulliken布居数为0.012,0.013;Ⅷ中O…H的Mulliken布居数为0.014,N…H的Mulliken布居数为0.031;Ⅸ中N…H的Mulliken布居数为0.019,0.019;Ⅹ中N…H的Mulliken布居数为0.012,0.012;Ⅺ中N…H的Mulliken布居数为0.004,O…H的Mulliken布居数为0.025。
下载:
导出CSV
| Bond | Overlap population | ||||
| Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ | |
| C(1)—C(2) | 0.541 | 0.540 | 0.537 | 0.539 | 0.530 |
| C(2)—C(3) | 0.529 | 0.529 | 0.530 | 0.528 | 0.526 |
| C(5)—C(7) | 0.272 | 0.270 | 0.271 | 0.264 | 0.269 |
| C(4)—O(13) | 0.273 | 0.274 | 0.261 | 0.273 | 0.272 |
| O(13)—C(8) | 0.273 | 0.273 | 0.266 | 0.273 | 0.273 |
| C(7)—C(8) | 0.458 | 0.455 | 0.458 | 0.457 | 0.455 |
| C(22)—C(23) | 0.538 | 0.540 | 0.537 | 0.536 | 0.538 |
| C(23)—C(24) | 0.527 | 0.528 | 0.530 | 0.532 | 0.527 |
| C(26)—C(28) | 0.272 | 0.271 | 0.271 | 0.273 | 0.271 |
| C(25)—O(34) | 0.257 | 0.264 | 0.261 | 0.243 | 0.255 |
| O(34)—C(29) | 0.265 | 0.255 | 0.266 | 0.260 | 0.265 |
| C(28)—C(29) | 0.456 | 0.462 | 0.458 | 0.458 | 0.455 |
| O(34)…H(14) | - | - | - | - | 0.008 |
| O(34)…H(16) | - | - | 0.017 | - | - |
| O(34)…H(17) | - | - | - | 0.014 | 0.008 |
| O(34)…H(18) | - | 0.007 | - | - | - |
| O(34)…H(19) | 0.007 | 0.008 | - | - | - |
| O(34)…H(20) | 0.007 | - | - | - | - |
| O(34)…H(21) | - | - | - | 0.014 | - |
| O(13)…H(37) | - | - | 0.017 | - | - |
下载:
导出CSV
| Bond | Overlap population | Bond | Overlap population | ||||
| Ⅵ | Ⅶ | Ⅷ | Ⅸ | Ⅹ | Ⅺ | ||
| C(1)—C(2) | 0.545 | 0.523 | 0.533 | 0.537 | 0.523 | C(1)—C(2) | 0.541 |
| C(2)—C(3) | 0.510 | 0.534 | 0.531 | 0.528 | 0.525 | C(2)—C(3) | 0.529 |
| C(5)—C(7) | 0.272 | 0.272 | 0.269 | 0.264 | 0.268 | C(5)—C(7) | 0.270 |
| C(4)—O(13) | 0.271 | 0.271 | 0.260 | 0.272 | 0.271 | C(4)—O(13) | 0.252 |
| O(13)—C(8) | 0.274 | 0.275 | 0.270 | 0.272 | 0.274 | O(13)—C(8) | 0.234 |
| C(7)—C(8) | 0.458 | 0.456 | 0.453 | 0.454 | 0.455 | C(7)—C(8) | 0.467 |
| C(22)—C(23) | 0.480 | 0.480 | 0.478 | 0.476 | 0.480 | C(23)—C(24) | 0.531 |
| C(23)—C(24) | 0.570 | 0.572 | 0.575 | 0.577 | 0.570 | C(24)—C(25) | 0.515 |
| C(24)—C(25) | 0.461 | 0.461 | 0.461 | 0.461 | 0.461 | C(25)—N(34) | 0.272 |
| C(25)—N(35) | 0.400 | 0.399 | 0.404 | 0.389 | 0.399 | C(29)—N(34) | 0.258 |
| C(29)—N(35) | 0.399 | 0.399 | 0.404 | 0.395 | 0.399 | C(26)—C(28) | 0.304 |
| C(28)—C(29) | 0.455 | 0.454 | 0.454 | 0.457 | 0.454 | C(28)—C(29) | 0.481 |
| N(35)…H(14) | 0.012 | - | - | - | 0.012 | N(34)…H(18) | 0.004 |
| N(35)…H(15) | 0.013 | 0.013 | - | - | - | O(13)…H(43) | 0.025 |
| N(35)…H(16) | - | 0.012 | 0.031 | - | - | - | - |
| N(35)…H(17) | - | - | - | 0.019 | 0.012 | - | - |
| N(35)…H(21) | - | - | - | 0.019 | - | - | - |
| O(13)…H(38) | - | - | 0.014 | - | - | - | - |
对照分子构型与原子间距(图 1、图 2和图 3)可见,在3组混合体系共11种构型中均有氢键的存在,且不同的构型有不同的氢键强度。第一组中,构型Ⅲ的Mulliken布居数较大;第二组中,构型Ⅷ的Mulliken布居数较大。表明这两种构型相互作用较明显,这与几何结构分析得到的结论相一致。
表 5列出了在M062X/6-31G(d)水平上混合体系的分子总能量、相互作用能、前线轨道能级及其差值。
下载:
导出CSV
| Energy | Etotal/Hartree | Δ E/(kJ/mol) | EHOMO/Hartree | ELUMO/Hartree | Δ EH-L/(kJ/mol) |
| YH | -537.107 | - | - | - | - |
| AD | -555.333 | - | - | - | - |
| KC | -517.252 | - | - | - | - |
| Ⅰ | -1074.219 | -14.668 | -0.267 | -0.004 | 688.774 |
| Ⅱ | -1074.219 | -14.840 | -0.266 | -0.005 | 686.122 |
| Ⅲ | -1074.218 | -12.125 | -0.269 | -0.002 | 699.092 |
| Ⅳ | -1074.221 | -18.541 | -0.266 | -0.006 | 682.709 |
| Ⅴ | -1074.219 | -14.620 | -0.266 | -0.005 | 684.284 |
| Ⅵ | -1092.447 | -20.480 | -0.260 | -0.048 | 555.477 |
| Ⅶ | -1092.447 | -21.033 | -0.259 | -0.047 | 555.425 |
| Ⅷ | -1092.446 | -17.796 | -0.257 | -0.046 | 555.556 |
| Ⅸ | -1092.449 | -24.497 | -0.259 | -0.049 | 551.539 |
| Ⅹ | -1092.447 | -21.047 | -0.260 | -0.048 | 555.451 |
| Ⅺ | -1054.369 | -27.357 | -0.242 | -0.011 | 605.939 |
由相互作用能ΔE可知,分子间的相互作用能在-12.125~-27.357 kJ/mol之间。第一组构型中,分子间相互作用能顺序为Ⅳ<Ⅱ<Ⅰ<Ⅴ<Ⅲ;第二组构型中,分子间相互作用能顺序为Ⅸ<Ⅹ<Ⅶ<Ⅵ<Ⅷ。其中,构型Ⅲ与构型Ⅷ的相互作用能较大,说明单体分子要形成混合体系Ⅲ和Ⅷ,只需要释放较少的能量,即体系Ⅲ和Ⅷ更易形成。
11种构型的占据轨道能量(EHOMO)均为负值,说明电子的状态比较稳定。第一组构型中,前线轨道能级差(ΔEHOMO-LUMO)的大小顺序为Ⅲ>Ⅰ>Ⅱ>Ⅴ>Ⅳ;第二组构型中,ΔEHOMO-LUMO大小顺序为Ⅷ>Ⅵ>Ⅹ>Ⅶ>Ⅸ。前线轨道能级差越大,分子构型也就越稳定,因为体系中电子的跃迁所需能量越大,跃迁越困难。所以,构型Ⅲ与构型Ⅷ的化学稳定性最好。
|
$ \Delta {\rm{E = E}}\left( {{\rm{AB}}} \right){\rm{-E}}\left( {\rm{A}} \right){\rm{-E}}\left( {\rm{B}} \right) $ |
为数据精确起见,在M062X/6-311++G(2d,2p)水平上对以上混合体系的分子总能量、相互作用能、前线轨道能级及其差值进行计算,得出表 6。
下载:
导出CSV
| Energy | Etotal/Hartree | Δ E/(kJ/mol) | EHOMO/Hartree | ELUMO/Hartree | Δ EH-L/(kJ/mol) |
| YH | -537.263 | - | - | - | - |
| AD | -555.493 | - | - | - | - |
| KC | -517.405 | - | - | - | - |
| Ⅰ | -1074.532 | -16.468 | -0.275 | -0.016 | 680.344 |
| Ⅱ | -1074.532 | -16.660 | -0.274 | -0.016 | 677.725 |
| Ⅲ | -1074.531 | -13.612 | -0.277 | -0.014 | 690.535 |
| Ⅳ | -1074.534 | -20.818 | -0.274 | -0.017 | 674.351 |
| Ⅴ | -1074.532 | -16.413 | -0.274 | -0.016 | 675.909 |
| Ⅵ | -1092.764 | -21.004 | -0.268 | -0.059 | 548.651 |
| Ⅶ | -1092.765 | -23.630 | -0.268 | -0.059 | 548.598 |
| Ⅷ | -1092.763 | -18.379 | -0.267 | -0.058 | 548.756 |
| Ⅸ | -1092.766 | -26.255 | -0.268 | -0.061 | 545.947 |
| Ⅹ | -1092.765 | -23.630 | -0.269 | -0.060 | 548.625 |
| Ⅺ | -1054.679 | -28.881 | -0.252 | -0.027 | 590.816 |
| Δ E=E(AB)- E(A)- E(B) | |||||
由表 6可知,分子间的相互作用能在-13.612~-28.881 kJ/mol之间,其中构型Ⅲ与构型Ⅷ的相互作用能较大,所以相比于其他构型,Ⅲ与Ⅷ更易于形成。
由前线轨道能级差(ΔEHOMO-LUMO)可知,构型Ⅲ与构型Ⅷ的能极差较大,所以化学稳定性较好。
综合考虑几何构型的原子间距、NBO电荷转移和Mulliken重叠布居,可得构型Ⅲ与构型Ⅷ的分子间相互作用较明显。
由表 7可见,沥青质分子Ⅰ在13种溶剂中的总溶剂化自由能(ΔGsolv)均为负值,表明Ⅰ分子经过溶剂化后能量降低,比溶剂化之前稳定。并且,远程静电部分的溶剂化自由能(ΔGelec)和短程非静电部分的溶剂化自由能(ΔGnonelec)也都为负值,说明沥青质分子Ⅰ受到的静电与非静电这两种作用都使溶质分子在加入溶剂后更趋于稳定,即远程静电作用与短程非静电作用同时存在于沥青质分子Ⅰ中的溶剂化过程中。
下载:
导出CSV
| 溶剂名称 | Δ Gelec/(kcal/mol) | Δ Gnonelec/(kcal/mol) | Δ Gsolv/(kcal/mol) |
| 乙腈 | -17.19 | -13.73 | -30.92 |
| 苯 | -6.85 | -23.51 | -30.36 |
| 氯苯 | -12.53 | -21.53 | -34.06 |
| 三氯甲烷 | -11.57 | -21.85 | -33.42 |
| 环己烷 | -5.94 | -20.80 | -26.74 |
| 二氯乙烷 | -14.76 | -17.40 | -32.16 |
| 二氯甲烷 | -14.35 | -20.73 | -35.08 |
| 二乙醚 | -11.00 | -19.47 | -30.47 |
| 乙醇 | -16.74 | -12.46 | -29.20 |
| 庚烷 | -5.51 | -22.21 | -27.72 |
| 甲醇 | -17.09 | -11.08 | -28.17 |
| 四氢呋喃 | -13.67 | -15.17 | -28.84 |
| 甲苯 | -7.19 | -22.52 | -29.71 |
其中,除乙腈、乙醇和甲醇外,非静电部分的溶剂化自由能的绝对值(|ΔGnonelec|)均明显大于静电部分的溶剂化自由能的绝对值(|ΔGelec|),说明与ΔGelec相比,ΔGnonelec对于ΔGsolv的贡献更为重要,并且不同溶剂中非静电部分溶剂化自由能ΔGnonelec值差别并不大。相反,不同溶剂中静电部分溶剂化自由能ΔGelec值差别却很大,从而造成总溶剂化自由能ΔGsolv在不同溶剂中差距较大。由此可以推断,在不同溶剂的沥青质溶剂化效应中,非静电作用差距不大,而静电作用差距较大。所以研究沥青质溶解性的关键可能在于远程静电作用的影响。
为了进一步研究沥青质分子Ⅰ在13种不同溶剂中的溶剂化自由能的变化,将这13种溶剂大体可以分为两类:第一类溶剂包括苯、氯苯、环己烷、二乙醚、正庚烷、四氢呋喃、甲苯等。其中正庚烷与环己烷为惰性溶剂,氯苯、二乙醚、四氢呋喃、甲苯这些溶剂是具有极性官能团的溶剂;第二类溶剂包括乙腈、二氯甲烷、氯仿、二氯乙烷、甲醇和乙醇,其中甲醇与乙醇属于质子型溶剂,乙腈、二氯甲烷和二氯乙烷分子中由于有吸电子基团的吸电子作用,所以存在显正电性的氢质子。
由表 7可知,与其他溶剂化过程相比,第二类溶剂的溶剂化过程中,总溶剂化自由能中非静电溶剂化自由能(ΔGnonelec)的绝对值较小,即所占比重普遍较小; 而静电溶剂化自由能(ΔGelec)的绝对值较大,其所占比重较大,说明沥青质分子Ⅰ与此类溶剂主要产生远程静电作用力。对于第一类溶剂的溶剂化过程,非静电溶剂化自由能(ΔGnonelec)所占比重普遍较大,而静电溶剂化自由能(ΔGelec)所占比重相对较小,表明在此类溶剂中溶剂与溶质主要产生短程非静电作用力。
观察总溶剂化自由能(ΔGsolv)可以看出,二氯甲烷、三氯甲烷(氯仿)、二氯乙烷、氯苯的溶剂化自由能的绝对值(|ΔGsolv|)较大,表明这四种溶剂对沥青质分子的溶解性较好,原因可能是这四种溶剂均含有强吸电子基团(如氯),以连续介质形态存在的大量溶剂分子由于这种强吸电子效应,使溶剂分子内和分子间存在电荷转移(即电子云变化),从而使溶剂极性变强或偶极矩变大,易与溶质分子的偶极矩发生远程静电作用。所以这几种溶剂的溶剂化效应中,ΔGelec所占比重较大,远程静电作用明显。这同样可以说明溶剂的远程静电作用的增强可能更利于沥青质分子的解聚与溶解,即溶解性的研究的关键在于静电部分。
乙腈、乙醇和甲醇与沥青质的溶剂化中,虽然静电部分(ΔGelec)作用较大,但由于非静电部分(ΔGnonelec)作用很小,导致总溶剂化自由能(ΔGsolv)并不大,溶解性一般。
根据表 7,正庚烷与环己烷对沥青质分子Ⅰ的溶剂化自由能的绝对值(|ΔGsolv|)最小,其非静电部分的作用强度一般,静电部分作用却很小。可能由于正庚烷和环己烷都是惰性溶剂,溶剂分子可与沥青质中的杂原子(O、N、S等)产生氢键作用,但由于两种溶剂的元素组成和空间结构使溶剂的偶极矩很小,无法与溶质分子的偶极矩产生较大静电作用。由此可说明脂肪烃类对沥青质的溶解性不佳,这也与实验相符合。
表 8为经简化后的沥青质分子Ⅲ在不同溶剂中的三种溶剂化自由能,可以得出与沥青质分子Ⅰ相类似的分析结果,例如,Ⅲ的不同溶剂化过程中,非静电部分溶剂化自由能(|ΔGnonelec|)多数大于静电部分的溶剂化自由能的绝对值(|ΔGelec|),即ΔGnonelec对于ΔGsolv的贡献更为重要;不同溶剂中静电部分溶剂化自由能ΔGelec值差别相对较大;二氯甲烷、三氯甲烷、二氯乙烷、氯苯对沥青质分子Ⅲ的溶解性较好,而脂肪烃对其溶解性较差。
下载:
导出CSV
| 溶剂名称 | ΔGelec/(kcal/mol) | ΔGnonelec/(kcal/mol) | ΔGsolv/(kcal/mol) |
| 乙腈 | -15.02 | -11.15 | -26.17 |
| 苯 | -6.06 | -19.29 | -25.35 |
| 氯苯 | -11.02 | -17.66 | -28.68 |
| 三氯甲烷 | -10.17 | -17.62 | -27.79 |
| 环己烷 | -5.27 | -17.06 | -22.33 |
| 二氯乙烷 | -12.94 | -14.08 | -27.02 |
| 二氯甲烷 | -12.59 | -16.81 | -29.40 |
| 二乙醚 | -9.70 | -16.01 | -25.71 |
| 乙醇 | -14.90 | -9.27 | -24.17 |
| 庚烷 | -4.89 | -18.22 | -23.11 |
| 甲醇 | -14.94 | -8.25 | -23.19 |
| 四氢呋喃 | -12.02 | -12.49 | -24.51 |
| 甲苯 | -6.42 | -18.51 | -24.93 |
由此可见,如分子Ⅰ与分子Ⅲ这样的以芳香分为核心,连以饱和环与烷基侧链的沥青质分子,其溶剂化研究与分析出的结论具有共性,也体现拥有这种结构特点的重质油沥青质分子性质相似。说明对于分子Ⅲ的简化合理。
运用密度泛函方法M062X计算得到了11种由沥青质分子片段——杂环分子(氧芴、吖啶、咔唑)组成的二元混合体系的全优化构型。通过NBO电荷分析得到混合体系中参与形成氢键的原子存在电荷转移,分子间也存在不同程度的定向电荷转移;由Mulliken重叠布居分析得出体系中的相互作用是由于氢键的形成;计算得到分子间的相互作用能在-13.612~-28.881 kJ/mol之间,并通过前线轨道能级差大小的比较得出了相对稳定的构型为Ⅲ与Ⅷ。在构型Ⅲ与Ⅷ中,杂原子均参与到了分子间氢键的形成,较为稳定。可知沥青质分子之间的相互作用与其杂原子的存在有关,这与石油沥青质聚沉的微观机理相一致[38]。
对沥青质模型分子在13种溶剂中进行溶剂化效应的建模和理论计算后,可得:(1)静电溶剂化自由能、非静电溶剂化自由能、总溶剂化自由能均为负值,说明静电与非静电作用都有助于沥青质的溶解;(2)沥青质溶剂化过程中的非静电作用差距不大,而静电作用差距较大,所以沥青质溶解性大小的关键可能在于溶剂对它的远程静电作用的大小;(3)二氯甲烷、氯仿、二氯乙烷、氯苯等对沥青质的溶解性较好,可能是由于溶剂中有强吸电子基团的存在,聚沉抑制剂的研究可参考此种类型的溶剂;正庚烷与环己烷等脂肪烃类对沥青质的溶解性效果不好。
本文通过沥青质片段分子间相互作用和溶剂化效应的建模与理论计算,对沥青质聚沉现象的微观机理进行了相关解释,并对聚沉的抑制作用进行了研究,为聚沉抑制剂的研发提供数据理论支持。
仁杰, 翁惠新, 刘馥英. 石油炼制与化工, 1993, (9):56~61. http://www.cnki.com.cn/Article/CJFDTotal-SYLH199309010.htm
G J Moridis, T A Blasingame, C M Freeman. Spe Latin American & Caribbean Petroleum Engineering Conference. Society of Petroleum Engineers, 2010.
张胜飞, 孙丽丽, 徐俊波 等. 物理化学学报, 2010, 26(1):57~65. http://www.cnki.com.cn/Article/CJFDTotal-WLHX201001010.htm
向柠, 王剑, 惠友权. 化学工业与工程技术, 2010, 31(4):25~30. http://www.cnki.com.cn/Article/CJFDTotal-HXGJ201004010.htm
T F Yen. Energy Sources, 1974, 1(4):447~463. doi: 10.1080/00908317408945937
L Robson, M Hunter. J. Membr. Biol., 2005, 204(1):39~47. doi: 10.1007/s00232-005-0745-8
C J Robinson, G L Cook. Anal. Chem., 1969, 41(12):1548~1554. doi: 10.1021/ac60281a005
邓文安, 阙国和. 石油学报(石油加工), 1997, 13(1):1~6. http://www.cnki.com.cn/article/cjfd1997-sxjg701.000.htm
J K Brown, W R Ladner. Fuel, 1960, 39(1):87~96.
Y Ruizmorales. J Phys. Chem. A, 2002, 106(46):11283~11308. doi: 10.1021/jp021152e
I García-Cruz, J M Martínez-Magadán, P Guadarrama et al. J. Phys. Chem. A, 2003, 107(10):1597~1603. doi: 10.1021/jp0216676
I García-Cruz, J M Martínez-Magadán, F Alvarez-Ramirez et al. J. Mol. Catal. A, 2005, 228(1~2):195-202. doi: 10.1016/j.molcata.2004.09.038
李能, 董明, 李龙 等. 石油沥青, 2014, 28(3):40~48. http://www.cnki.com.cn/Article/CJFDTotal-OILE201403019.htm
王春璐, 周涵, 王子军 等. 计算机与应用化学, 2012, 29(10):1221~1224. http://www.cnki.com.cn/Article/CJFDTotal-JSYH201210017.htm
孙昱东, 杨朝合, 山红红 等. 石油学报(石油加工), 2010, 26(1):191~197. http://www.cnki.com.cn/Article/CJFDTotal-SXJG2010S1039.htm
Y D Sun, C H Yang, H Zhao et al. Energy Fuels, 2010, 24(9):5008~5011. doi: 10.1021/ef1005385
S F R A ultanov, Y A Tileuberdi, Y K A Ongarbayev et al. Eurasian Chem. Technol. J., 2012, 15(1):77~81. doi: 10.18321/ectj143
王跃, 张会成, 凌凤香 等. 石油炼制与化工, 2012, 43(7):52~56. http://www.cnki.com.cn/Article/CJFDTotal-SYLH201207018.htm
H Groenzin, O C Mullins. Energy Fuels, 2000, 14(3):677~684. doi: 10.1021/ef990225z
胡玉峰, 杨兰英, 林雄森 等. 石油勘探与开发, 2000, 27(5):109~111. http://www.cnki.com.cn/Article/CJFDTotal-SKYK200005033.htm
赵凤兰, 鄢捷年. 油田化学, 2004, 21(4):310~312. http://www.cnki.com.cn/Article/CJFDTotal-YJHX200404005.htm
何行范, 李士伦. 天然气工业, 2003, 23(2):78~81. http://www.cnki.com.cn/Article/CJFDTotal-TRQG200302030.htm
陈欢庆, 曹晨, 梁淑贤 等. 天然气地球科学, 2013, 24(2):227~237. http://www.cnki.com.cn/Article/CJFDTotal-TDKX201302006.htm
赖锦, 王贵文, 孟辰卿 等. 地球物理学进展, 2015, 30(1):217~227. http://www.cnki.com.cn/Article/CJFDTotal-DQWJ201501033.htm
T Takanohashi, S Sato, R P Tanaka. Liq. Fuels Technol., 2003, 21(3~4):491-505.
E Rogel. Colloids Surf. A, 1995, 104(1):85~93. doi: 10.1016/0927-7757(95)03234-5
E Rogel. Energy Fuels, 2000, 14(3):566~574. doi: 10.1021/ef990166p
E G Hohenstein, S T Chill, C D Sherrill. J. Chem. Theory Comput., 2008, 4(12):1996~2000. doi: 10.1021/ct800308k
袁煙, 吕玲玲, 王云普 等. 科学通报, 2007, 52(10):1128~1135. http://www.cnki.com.cn/Article/CJFDTotal-KXTB200710007.htm
孙涛, 王一波. 物理化学学报, 2011, 27(11):2553~2558. http://www.cnki.com.cn/Article/CJFDTotal-WLHX201111011.htm
孙如. 苏州大学博士学位论文, 2008.
谷开慧. 东北师范大学硕士学位论文, 2007.
A D Becke. J. Chem. Phys., 1993, 98(2):1372~1377. doi: 10.1063/1.464304
M J Frisch, G W Trucks, H B Schlegel et al. Gaussian 09, Gaussian, Inc. 2009.
I Mayer, P Salvador. Chem. Phys. Lett., 2004, 383(3):368~375.
A Grechnev, R Ahuja, O Eriksson. J. Phys.:Condens. Matter, 2005, 15(45):7751~7761.
J Oláh, F Blockhuys, T Veszprémi et al. Eur. J. Inorg. Chem., 2006, 2006(1):69~77.
卢贵武, 李英峰, 宋辉 等. 石油勘探与开发, 2008, 35(1):67~72. http://www.cnki.com.cn/Article/CJFDTotal-SKYK200801015.htm

图式 2 经Ⅱ简化后的沥青质模型分子Ⅲ
Scheme 2 Asphaltene model molecule Ⅲ simplified by model molecule Ⅱ

图 1 氧芴(YH)及YH+YH优化后的几何结构、原子编号及分子间距
Figure 1 Atomic number, optimized geometries and intermolecular distance(Å) of dibenzofuran(YH)and YH+YH

图 2 氧芴(YH),吖啶(AD)及YH+AD优化后的几何结构、原子编号及分子间距
Figure 2 Atomic number, optimized geometries and intermolecular distance(Å) of dibenzofuran(YH), Acridine(AD)and YH+AD

图 3 氧芴(YH),咔唑(KC)及YH+KC优化后的几何结构、原子编号及分子间距
Figure 3 Atomic number, optimized geometries and intermolecular distance(Å) of dibenzofuran(YH), Carbazole (KC)and YH+KC
表 1 氧芴(YH),氧芴+氧芴(YH+YH)的NBO电荷(e)
Table 1. Natural atomic charges(e) of dibenzofuran(YH) and YH+YH
| Atom | Isolated molecule | Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ |
| C(1) | -0.259 | -0.259 | -0.259 | -0.258 | -0.257 | -0.267 |
| C(2) | -0.231 | -0.231 | -0.231 | -0.230 | -0.231 | -0.231 |
| C(3) | -0.281 | -0.281 | -0.281 | -0.280 | -0.282 | -0.282 |
| C(4) | 0.333 | 0.332 | 0.332 | 0.331 | 0.331 | 0.331 |
| C(5) | -0.112 | -0.112 | -0.112 | -0.113 | -0.118 | -0.113 |
| C(6) | -0.210 | -0.210 | -0.210 | -0.211 | -0.215 | -0.215 |
| C(7) | -0.112 | -0.114 | -0.114 | -0.110 | -0.119 | -0.112 |
| C(8) | 0.333 | 0.331 | 0.331 | 0.332 | 0.331 | 0.331 |
| C(9) | -0.281 | -0.281 | -0.288 | -0.281 | -0.282 | -0.281 |
| C(10) | -0.231 | -0.237 | -0.236 | -0.231 | -0.231 | -0.231 |
| C(11) | -0.259 | -0.267 | -0.259 | -0.258 | -0.258 | -0.259 |
| C(12) | -0.210 | -0.211 | -0.211 | -0.210 | -0.216 | -0.209 |
| O(13) | -0.474 | -0.475 | -0.474 | -0.485 | -0.475 | -0.475 |
| H(14) | 0.246 | 0.246 | 0.246 | 0.246 | 0.246 | 0.255 |
| H(15) | 0.247 | 0.247 | 0.247 | 0.245 | 0.246 | 0.247 |
| H(16) | 0.258 | 0.258 | 0.258 | 0.272 | 0.257 | 0.257 |
| H(17) | 0.246 | 0.246 | 0.246 | 0.246 | 0.258 | 0.254 |
| H(18) | 0.258 | 0.258 | 0.265 | 0.255 | 0.257 | 0.257 |
| H(19) | 0.247 | 0.256 | 0.254 | 0.247 | 0.246 | 0.246 |
| H(20) | 0.246 | 0.256 | 0.246 | 0.246 | 0.246 | 0.246 |
| H(21) | 0.246 | 0.246 | 0.246 | 0.247 | 0.258 | 0.248 |
| C(22) | -0.259 | -0.257 | -0.257 | -0.258 | -0.256 | -0.257 |
| C(23) | -0.231 | -0.228 | -0.230 | -0.230 | -0.226 | -0.228 |
| C(24) | -0.281 | -0.279 | -0.280 | -0.280 | -0.275 | -0.280 |
| C(25) | 0.333 | 0.330 | 0.332 | 0.331 | 0.328 | 0.329 |
| C(26) | -0.112 | -0.112 | -0.111 | -0.113 | -0.111 | -0.112 |
| C(27) | -0.210 | -0.211 | -0.210 | -0.211 | -0.211 | -0.211 |
| C(28) | -0.112 | -0.111 | -0.112 | -0.110 | -0.109 | -0.111 |
| C(29) | 0.333 | 0.331 | 0.330 | 0.332 | 0.330 | 0.331 |
| C(30) | -0.281 | -0.281 | -0.280 | -0.281 | -0.281 | -0.281 |
| C(31) | -0.231 | -0.230 | -0.228 | -0.231 | -0.230 | -0.230 |
| C(32) | -0.259 | -0.258 | -0.257 | -0.258 | -0.257 | -0.257 |
| C(33) | -0.210 | -0.210 | -0.210 | -0.210 | -0.210 | -0.210 |
| O(34) | -0.474 | -0.488 | -0.490 | -0.485 | -0.493 | -0.489 |
| H(35) | 0.246 | 0.246 | 0.247 | 0.246 | 0.247 | 0.247 |
| H(36) | 0.247 | 0.248 | 0.248 | 0.245 | 0.250 | 0.249 |
| H(37) | 0.258 | 0.266 | 0.259 | 0.272 | 0.264 | 0.265 |
| H(38) | 0.246 | 0.246 | 0.247 | 0.246 | 0.247 | 0.247 |
| H(39) | 0.258 | 0.258 | 0.264 | 0.255 | 0.259 | 0.259 |
| H(40) | 0.247 | 0.248 | 0.248 | 0.247 | 0.248 | 0.248 |
| H(41) | 0.246 | 0.247 | 0.247 | 0.246 | 0.247 | 0.247 |
| H(42) | 0.246 | 0.247 | 0.247 | 0.247 | 0.247 | 0.247 |
 下载: 导出CSV
下载: 导出CSV
表 2 氧芴(YH)、吖啶(AD)、咔唑(KC)、氧芴+吖啶(YH+AD)、氧芴+咔唑(YH+KC)的NBO电荷(e)
Table 2. Natural atomic charges(e) of dibenzofuran(YH), acridine(AD), carbazole (KC), YH+AD and YH+KC
| Atom | Isolated | Ⅵ | Ⅶ | Ⅷ | Ⅸ | Ⅹ | Atom | Isolated | Ⅺ |
| molecule | molecule | ||||||||
| C(1) | -0.259 | -0.263 | -0.260 | -0.258 | -0.260 | -0.263 | C(1) | -0.259 | -0.255 |
| C(2) | -0.231 | -0.235 | -0.234 | -0.233 | -0.232 | -0.231 | C(2) | -0.231 | -0.228 |
| C(3) | -0.281 | -0.283 | -0.286 | -0.281 | -0.284 | -0.283 | C(3) | -0.281 | -0.281 |
| C(4) | 0.333 | 0.330 | 0.330 | 0.331 | 0.330 | 0.329 | C(4) | 0.333 | 0.330 |
| C(5) | -0.112 | -0.115 | -0.116 | -0.114 | -0.117 | -0.115 | C(5) | -0.112 | -0.108 |
| C(6) | -0.210 | -0.212 | -0.212 | -0.214 | -0.214 | -0.216 | C(6) | -0.210 | -0.208 |
| C(7) | -0.112 | -0.111 | -0.112 | -0.110 | -0.117 | -0.111 | C(7) | -0.112 | -0.110 |
| C(8) | 0.333 | 0.331 | 0.332 | 0.333 | 0.330 | 0.331 | C(8) | 0.333 | 0.325 |
| C(9) | -0.281 | -0.281 | -0.281 | -0.282 | -0.284 | -0.282 | C(9) | -0.281 | -0.278 |
| C(10) | -0.231 | -0.233 | -0.233 | -0.232 | -0.232 | -0.233 | C(10) | -0.231 | -0.225 |
| C(11) | -0.259 | -0.260 | -0.260 | -0.259 | -0.260 | -0.260 | C(11) | -0.259 | -0.254 |
| C(12) | -0.210 | -0.211 | -0.211 | -0.210 | -0.214 | -0.210 | C(12) | -0.210 | -0.210 |
| O(13) | -0.474 | -0.476 | -0.475 | -0.483 | -0.477 | -0.476 | O(13) | -0.474 | -0.509 |
| H(14) | 0.246 | 0.258 | 0.244 | 0.244 | 0.245 | 0.259 | H(14) | 0.246 | 0.248 |
| H(15) | 0.247 | 0.261 | 0.259 | 0.243 | 0.245 | 0.245 | H(15) | 0.247 | 0.250 |
| H(16) | 0.258 | 0.256 | 0.271 | 0.280 | 0.256 | 0.256 | H(16) | 0.258 | 0.259 |
| H(17) | 0.246 | 0.244 | 0.244 | 0.245 | 0.263 | 0.260 | H(17) | 0.246 | 0.249 |
| H(18) | 0.258 | 0.257 | 0.257 | 0.255 | 0.256 | 0.257 | H(18) | 0.258 | 0.269 |
| H(19) | 0.247 | 0.246 | 0.246 | 0.246 | 0.245 | 0.246 | H(19) | 0.247 | 0.251 |
| H(20) | 0.246 | 0.245 | 0.245 | 0.245 | 0.245 | 0.245 | H(20) | 0.246 | 0.248 |
| H(21) | 0.246 | 0.246 | 0.245 | 0.246 | 0.263 | 0.248 | H(21) | 0.246 | 0.248 |
| C(22) | -0.240 | -0.239 | -0.239 | -0.238 | -0.237 | -0.239 | C(22) | -0.270 | -0.271 |
| C(23) | -0.235 | -0.231 | -0.231 | -0.232 | -0.227 | -0.231 | C(23) | -0.231 | -0.232 |
| C(24) | -0.221 | -0.223 | -0.223 | -0.222 | -0.219 | -0.224 | C(24) | -0.279 | -0.281 |
| C(25) | 0.191 | 0.195 | 0.195 | 0.196 | 0.197 | 0.195 | C(25) | 0.181 | 0.173 |
| C(26) | -0.104 | -0.104 | -0.104 | -0.103 | -0.103 | -0.104 | C(26) | -0.093 | -0.095 |
| C(27) | -0.212 | -0.211 | -0.211 | -0.213 | -0.212 | -0.211 | C(27) | -0.206 | -0.208 |
| C(28) | -0.104 | -0.104 | -0.104 | -0.104 | -0.105 | -0.104 | C(28) | -0.093 | -0.094 |
| C(29) | 0.191 | 0.195 | 0.195 | 0.195 | 0.195 | 0.195 | C(29) | 0.181 | 0.180 |
| C(30) | -0.221 | -0.223 | -0.223 | -0.223 | -0.225 | -0.224 | C(30) | -0.279 | -0.278 |
| C(31) | -0.235 | -0.231 | -0.231 | -0.232 | -0.230 | -0.231 | C(31) | -0.231 | -0.233 |
| C(32) | -0.240 | -0.239 | -0.239 | -0.240 | -0.240 | -0.239 | C(32) | -0.270 | -0.271 |
| C(33) | -0.212 | -0.211 | -0.211 | -0.210 | -0.210 | -0.211 | C(33) | -0.206 | -0.207 |
| C(34) | -0.144 | -0.140 | -0.140 | -0.140 | -0.138 | -0.140 | N(34) | -0.585 | -0.594 |
| N(35) | -0.438 | -0.465 | -0.464 | -0.458 | -0.471 | -0.466 | H(35) | 0.243 | 0.242 |
| H(36) | 0.246 | 0.247 | 0.247 | 0.246 | 0.247 | 0.247 | H(36) | 0.243 | 0.243 |
| H(37) | 0.247 | 0.248 | 0.249 | 0.247 | 0.250 | 0.249 | H(37) | 0.242 | 0.250 |
| H(38) | 0.256 | 0.258 | 0.259 | 0.265 | 0.258 | 0.258 | H(38) | 0.243 | 0.242 |
| H(39) | 0.242 | 0.243 | 0.243 | 0.242 | 0.243 | 0.243 | H(39) | 0.242 | 0.243 |
| H(40) | 0.256 | 0.259 | 0.258 | 0.252 | 0.256 | 0.259 | H(40) | 0.243 | 0.242 |
| H(41) | 0.247 | 0.249 | 0.248 | 0.247 | 0.249 | 0.249 | H(41) | 0.243 | 0.242 |
| H(42) | 0.246 | 0.247 | 0.247 | 0.247 | 0.248 | 0.247 | H(42) | 0.243 | 0.242 |
| H(43) | 0.242 | 0.243 | 0.243 | 0.243 | 0.243 | 0.243 | H(43) | 0.439 | 0.454 |
| H(44) | 0.242 | 0.243 | 0.243 | 0.243 | 0.244 | 0.243 |
下载: 导出CSV
表 3 氧芴+氧芴(YH+YH)的重叠布居分析结果
Table 3. Overlap population of YH+YH
| Bond | Overlap population | ||||
| Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ | |
| C(1)—C(2) | 0.541 | 0.540 | 0.537 | 0.539 | 0.530 |
| C(2)—C(3) | 0.529 | 0.529 | 0.530 | 0.528 | 0.526 |
| C(5)—C(7) | 0.272 | 0.270 | 0.271 | 0.264 | 0.269 |
| C(4)—O(13) | 0.273 | 0.274 | 0.261 | 0.273 | 0.272 |
| O(13)—C(8) | 0.273 | 0.273 | 0.266 | 0.273 | 0.273 |
| C(7)—C(8) | 0.458 | 0.455 | 0.458 | 0.457 | 0.455 |
| C(22)—C(23) | 0.538 | 0.540 | 0.537 | 0.536 | 0.538 |
| C(23)—C(24) | 0.527 | 0.528 | 0.530 | 0.532 | 0.527 |
| C(26)—C(28) | 0.272 | 0.271 | 0.271 | 0.273 | 0.271 |
| C(25)—O(34) | 0.257 | 0.264 | 0.261 | 0.243 | 0.255 |
| O(34)—C(29) | 0.265 | 0.255 | 0.266 | 0.260 | 0.265 |
| C(28)—C(29) | 0.456 | 0.462 | 0.458 | 0.458 | 0.455 |
| O(34)…H(14) | - | - | - | - | 0.008 |
| O(34)…H(16) | - | - | 0.017 | - | - |
| O(34)…H(17) | - | - | - | 0.014 | 0.008 |
| O(34)…H(18) | - | 0.007 | - | - | - |
| O(34)…H(19) | 0.007 | 0.008 | - | - | - |
| O(34)…H(20) | 0.007 | - | - | - | - |
| O(34)…H(21) | - | - | - | 0.014 | - |
| O(13)…H(37) | - | - | 0.017 | - | - |
下载: 导出CSV
表 4 氧芴+吖啶(YH+AD)、氧芴+咔唑(YH+KC)的重叠布居分析结果
Table 4. Overlap population of YH+AD and YH+KC
| Bond | Overlap population | Bond | Overlap population | ||||
| Ⅵ | Ⅶ | Ⅷ | Ⅸ | Ⅹ | Ⅺ | ||
| C(1)—C(2) | 0.545 | 0.523 | 0.533 | 0.537 | 0.523 | C(1)—C(2) | 0.541 |
| C(2)—C(3) | 0.510 | 0.534 | 0.531 | 0.528 | 0.525 | C(2)—C(3) | 0.529 |
| C(5)—C(7) | 0.272 | 0.272 | 0.269 | 0.264 | 0.268 | C(5)—C(7) | 0.270 |
| C(4)—O(13) | 0.271 | 0.271 | 0.260 | 0.272 | 0.271 | C(4)—O(13) | 0.252 |
| O(13)—C(8) | 0.274 | 0.275 | 0.270 | 0.272 | 0.274 | O(13)—C(8) | 0.234 |
| C(7)—C(8) | 0.458 | 0.456 | 0.453 | 0.454 | 0.455 | C(7)—C(8) | 0.467 |
| C(22)—C(23) | 0.480 | 0.480 | 0.478 | 0.476 | 0.480 | C(23)—C(24) | 0.531 |
| C(23)—C(24) | 0.570 | 0.572 | 0.575 | 0.577 | 0.570 | C(24)—C(25) | 0.515 |
| C(24)—C(25) | 0.461 | 0.461 | 0.461 | 0.461 | 0.461 | C(25)—N(34) | 0.272 |
| C(25)—N(35) | 0.400 | 0.399 | 0.404 | 0.389 | 0.399 | C(29)—N(34) | 0.258 |
| C(29)—N(35) | 0.399 | 0.399 | 0.404 | 0.395 | 0.399 | C(26)—C(28) | 0.304 |
| C(28)—C(29) | 0.455 | 0.454 | 0.454 | 0.457 | 0.454 | C(28)—C(29) | 0.481 |
| N(35)…H(14) | 0.012 | - | - | - | 0.012 | N(34)…H(18) | 0.004 |
| N(35)…H(15) | 0.013 | 0.013 | - | - | - | O(13)…H(43) | 0.025 |
| N(35)…H(16) | - | 0.012 | 0.031 | - | - | - | - |
| N(35)…H(17) | - | - | - | 0.019 | 0.012 | - | - |
| N(35)…H(21) | - | - | - | 0.019 | - | - | - |
| O(13)…H(38) | - | - | 0.014 | - | - | - | - |
下载: 导出CSV
表 5 M062X/6-31G(d)水平的分子总能量、分子间作用能以及前线轨道能级差
Table 5. Total energy, interaction energy, frontier orbital energies and ΔEHOMO-LUMO (kJ/mol) at the M062X/6-31G(d) level
| Energy | Etotal/Hartree | Δ E/(kJ/mol) | EHOMO/Hartree | ELUMO/Hartree | Δ EH-L/(kJ/mol) |
| YH | -537.107 | - | - | - | - |
| AD | -555.333 | - | - | - | - |
| KC | -517.252 | - | - | - | - |
| Ⅰ | -1074.219 | -14.668 | -0.267 | -0.004 | 688.774 |
| Ⅱ | -1074.219 | -14.840 | -0.266 | -0.005 | 686.122 |
| Ⅲ | -1074.218 | -12.125 | -0.269 | -0.002 | 699.092 |
| Ⅳ | -1074.221 | -18.541 | -0.266 | -0.006 | 682.709 |
| Ⅴ | -1074.219 | -14.620 | -0.266 | -0.005 | 684.284 |
| Ⅵ | -1092.447 | -20.480 | -0.260 | -0.048 | 555.477 |
| Ⅶ | -1092.447 | -21.033 | -0.259 | -0.047 | 555.425 |
| Ⅷ | -1092.446 | -17.796 | -0.257 | -0.046 | 555.556 |
| Ⅸ | -1092.449 | -24.497 | -0.259 | -0.049 | 551.539 |
| Ⅹ | -1092.447 | -21.047 | -0.260 | -0.048 | 555.451 |
| Ⅺ | -1054.369 | -27.357 | -0.242 | -0.011 | 605.939 |
下载: 导出CSV
表 6 M062X/6-311++G(2d,2p)水平的分子总能量、分子间作用能以及前线轨道能级差
Table 6. Total energy, interaction energy, frontier orbital energies and ΔEHOMO-LUMO (kJ/mol) at the M062X/6-311++G(2d, 2p) level
| Energy | Etotal/Hartree | Δ E/(kJ/mol) | EHOMO/Hartree | ELUMO/Hartree | Δ EH-L/(kJ/mol) |
| YH | -537.263 | - | - | - | - |
| AD | -555.493 | - | - | - | - |
| KC | -517.405 | - | - | - | - |
| Ⅰ | -1074.532 | -16.468 | -0.275 | -0.016 | 680.344 |
| Ⅱ | -1074.532 | -16.660 | -0.274 | -0.016 | 677.725 |
| Ⅲ | -1074.531 | -13.612 | -0.277 | -0.014 | 690.535 |
| Ⅳ | -1074.534 | -20.818 | -0.274 | -0.017 | 674.351 |
| Ⅴ | -1074.532 | -16.413 | -0.274 | -0.016 | 675.909 |
| Ⅵ | -1092.764 | -21.004 | -0.268 | -0.059 | 548.651 |
| Ⅶ | -1092.765 | -23.630 | -0.268 | -0.059 | 548.598 |
| Ⅷ | -1092.763 | -18.379 | -0.267 | -0.058 | 548.756 |
| Ⅸ | -1092.766 | -26.255 | -0.268 | -0.061 | 545.947 |
| Ⅹ | -1092.765 | -23.630 | -0.269 | -0.060 | 548.625 |
| Ⅺ | -1054.679 | -28.881 | -0.252 | -0.027 | 590.816 |
| Δ E=E(AB)- E(A)- E(B) | |||||
下载: 导出CSV
表 7 沥青质分子Ⅰ在13种溶剂中的静电(ΔGelec)、非静电(ΔGnonelec)及总溶剂化自由能(ΔGsolv)
Table 7. ΔGelec, ΔGnonelec and ΔGsolv of asphaltene molecules I in 13 solvents
| 溶剂名称 | Δ Gelec/(kcal/mol) | Δ Gnonelec/(kcal/mol) | Δ Gsolv/(kcal/mol) |
| 乙腈 | -17.19 | -13.73 | -30.92 |
| 苯 | -6.85 | -23.51 | -30.36 |
| 氯苯 | -12.53 | -21.53 | -34.06 |
| 三氯甲烷 | -11.57 | -21.85 | -33.42 |
| 环己烷 | -5.94 | -20.80 | -26.74 |
| 二氯乙烷 | -14.76 | -17.40 | -32.16 |
| 二氯甲烷 | -14.35 | -20.73 | -35.08 |
| 二乙醚 | -11.00 | -19.47 | -30.47 |
| 乙醇 | -16.74 | -12.46 | -29.20 |
| 庚烷 | -5.51 | -22.21 | -27.72 |
| 甲醇 | -17.09 | -11.08 | -28.17 |
| 四氢呋喃 | -13.67 | -15.17 | -28.84 |
| 甲苯 | -7.19 | -22.52 | -29.71 |
下载: 导出CSV
表 8 沥青质分子Ⅲ在13种溶剂中的静电(ΔGelec)、非静电(ΔGnonelec)及总溶剂化自由能(ΔGsolv)
Table 8. ΔGelec, ΔGnonelec and ΔGsolv of asphaltene molecules Ⅲ in 13 solvents
| 溶剂名称 | ΔGelec/(kcal/mol) | ΔGnonelec/(kcal/mol) | ΔGsolv/(kcal/mol) |
| 乙腈 | -15.02 | -11.15 | -26.17 |
| 苯 | -6.06 | -19.29 | -25.35 |
| 氯苯 | -11.02 | -17.66 | -28.68 |
| 三氯甲烷 | -10.17 | -17.62 | -27.79 |
| 环己烷 | -5.27 | -17.06 | -22.33 |
| 二氯乙烷 | -12.94 | -14.08 | -27.02 |
| 二氯甲烷 | -12.59 | -16.81 | -29.40 |
| 二乙醚 | -9.70 | -16.01 | -25.71 |
| 乙醇 | -14.90 | -9.27 | -24.17 |
| 庚烷 | -4.89 | -18.22 | -23.11 |
| 甲醇 | -14.94 | -8.25 | -23.19 |
| 四氢呋喃 | -12.02 | -12.49 | -24.51 |
| 甲苯 | -6.42 | -18.51 | -24.93 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
