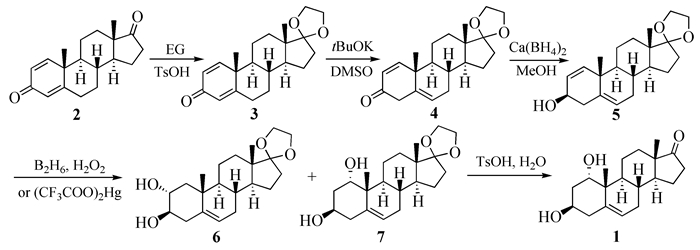
图式 1.
Kaneko等报道的1的合成路线
Scheme 1.
Synthetic route reported by Kaneko et al

1α-羟基去氢表雄酮属于甾体类化合物,它是合成马沙骨化醇、他卡西醇、阿法骨化醇、氟骨三醇等活性维生素D3类药物的关键中间体[1],也可用于合成其他天然产物如(+)-Withanolide E[2]、Sominone及其衍生物[3]、Cyclocitrinols[4]等,同时它也是合成维生素D衍生物的重要原料[5]。不过,由于立体选择性和区域选择性地引入1α羟基相当具有挑战性,因此其合成条件苛刻、收率较低,1α-羟基去氢表雄酮原料匮乏,一定程度制约了活性维生素D类药物的生产、研发。因此,开发高效经济、技术可行、安全环保、适于生产的1α-羟基去氢表雄酮合成工艺具有重要现实意义和广阔应用前景。
目前,文献报道的1α-羟基去氢表雄酮(化合物1,结构见图式 1)生产方法有化学合成法和生物转化法[6]。1973年,Kaneko等[7]以1, 4-雄二烯二酮为原料,经羰基保护、碱催化的烯键异构、Ca(BH4)2还原、硼氢化氧化制得2α, 3β-二羟基(化合物6,20%)和lα, 3β-二羟基(化合物7,15%)两种构型产物。5-位双键的存在和还原反应缺乏选择性使得羟化产物较为复杂。经柱色谱分离得到化合物7,再在酸性条件下脱去保护基,最终制得1。由于收率较低(整条路线收率小于7%)且使用毒性较大的乙硼烷,故该路线不适用于工业化生产。Hiroyuki等[8]将中间体5经三氟乙酸汞处理、硼氢化钠还原得中间体7,其单步收率为36.5%,但是使用了三氟乙酸汞,也不适于工业化生产。2007年,Sheikh等[4]在合成天然化合物Cyclocitrinols的过程中合成了乙酰基保护的1。以去氢表雄酮为原料,经17-位羰基保护、二氯二氰基苯醌(DDQ)脱氢、碱性双氧水环氧化、液氨电子还原后,脱去缩酮保护基得到1,总收率为21%。虽然该方法较之前的报道在收率上有很大的提高,但是DDQ氧化反应收率较低(57%)且后处理困难,故该路线也不适于量产。针对DDQ脱氢收率低和后处理困难等不足,刘兆鹏等[9]对该工艺路线进行了改进。在烯丙基磷酸二乙酯存在下,利用钯碳催化保护的去氢表雄酮脱氢得到中间体8,单步收率达89%。但使用烯丙基磷酸二乙酯的量较大,是底物的6倍,生产成本较高,液氨-锂还原也是在-40~-80℃下进行,且产物需柱色谱纯化,因此该方法还需进一步优化。最近笔者课题组在以上合成路线的基础上进行了改进[10]:以1, 4-雄二烯二酮为原料,经羰基保护、溴代、异构化、还原、环氧化、开环和水解7步反应以28.3%的收率得到1α-羟基去氢表雄酮。其中,中间体5经Sharpless环氧化引入lα, 2α-环氧环,再还原引入1α-羟基。由于环氧化步骤收率不高且需要柱色谱分离,因此该方法仍需优化。
此外,通过微生物合成1的探索已经进行了半个多世纪。1960年,Dodson等[11]公开了通过微生物合成1的方法,以去氢表雄酮为原料,菌种采用Penicillium.sp ATCC 12556,液体发酵培养24h,粗品收率约为33.3%,且需要柱色谱进行分离,生产成本较高。Fujiwara等[12]采用菌种Penicillium Oxalicum IFO-7000合成化合物1,转化率为45.0%,但转化效率不高,每升培养基只能转化得到7g产物,这使得大量生产较为困难。2010年,刘佩卉等[13]等用斜卧青霉(Penicillium decumbens)UC086将去氢表雄酮转化为1,转化率为18.83%,但每升培养基只能转化6g去氢表雄酮。生物合成法生产周期长、生产成本高、转化效率低,后续处理多以有机溶剂萃取、柱色谱纯化产物,这使其的应用受到限制。对于大规模生产而言,还需对生物转化法进行深入研究以适应工业化生产的要求。
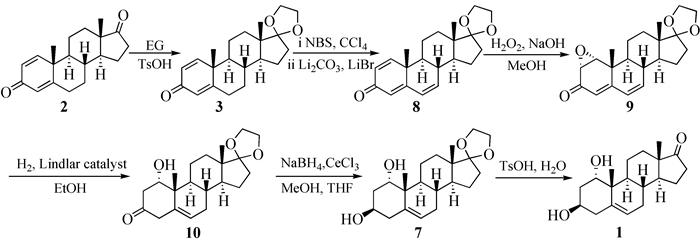
通过上述回顾,可以看到国内外主要依靠微生物发酵制备1α-羟基去氢表雄酮,这远不能满足市场需求;另一方面,微生物后处理困难,需要提取与柱层析等复杂的过程,这些也给大规模生产造成困难。因此,探索一条原料易得、操作简便、条件温和、适于生产的合成路线显得尤为重要。本文以1, 4-雄烯二酮为原料,经羰基保护、消除、环氧化、还原和水解等6步反应以29%总收率合成1α-羟基去氢表雄酮。该合成路线(图式 2)避免使用DDQ等昂贵试剂和液氨-锂等苛刻条件,条件温和、安全环保、较适于生产。
熔点用X4型显微熔点仪测定(温度未校正);核磁共振谱仪为Brucker Avance-400型,TMS为内标;质谱仪为LTQ-Orbitrap XL型(Thermo Fisher),离子源采用电喷雾离子化(ESI);旋光仪为BWXG4型圆盘旋光仪;柱色谱使用200~300目硅胶(购自青岛海洋化工厂);薄层色谱使用Silica Gel GF254(购自青岛海洋化工厂);所有试剂均为国产或进口分析纯,无水溶剂用常规方法干燥处理。
向装有分水器的250mL反应瓶中加入7.11g(25mmol)1, 4-雄烯二酮、3.1g (50mmol)乙二醇、100mg对甲苯磺酸和100mL甲苯,加热回流反应12h,TLC检测至反应完全。反应液冷至室温,用100mL饱和碳酸氢钠水溶液和乙酸乙酯(50mL×3)萃取,合并有机层,无水硫酸镁干燥,减压浓缩。乙酸乙酯结晶,得到类白色固体粉末7.9g,收率97%,熔点170~171℃(文献值[14]171~172℃)。1H NMR (400MHz,CDCl3) δ:7.06 (d,J=10.1Hz,1H),6.23(dd,J=10.1、1.9 Hz,1H),6.07 (t,J=1.5Hz,1H),3.93~3.80(m,4H),2.52~2.41(m,1H),2.40~2.32 (m,1H),2.05~1.93(m,2H),1.85~1.74(m,2H),1.73~1.53(m,4H),1.48~1.26(m,3H),1.23(s,3H),1.14~1.00(m,2H),0.92(s,3H);13C NMR (101MHz,CDCl3)δ:186.37,169.15,155.84,127.48,123.86,118.93,65.24,64.55,52.16,49.30,45.94,43.58,35.81,34.03,32.91,32.79,30.24,22.78,22.30,18.73,14.37;ESI-MS m/z:329.5 [M+H]+。
向250mL反应瓶中加入6.56g(20mmol)上述合成的化合物3、4.27g(24mmol)N-溴代丁二酰亚胺、100mg偶氮二异丁腈和100mL四氯化碳,回流反应4h,TLC监测至反应完全。反应液降至室温,抽滤除去固体不溶物,滤饼用四氯化碳洗涤,滤液减压浓缩至干。该溴代产物不经纯化直接使用。向残留物中加入1.92g(22mmol)溴化锂、1.63g(22mmol)碳酸锂和100mL DMF,加热至100℃,搅拌反应2h,TLC监测反应完全。反应液抽滤除去固体,滤液减压浓缩。粗产物经乙酸乙酯结晶,得到类白色固体粉末5.68g,两步收率共计87%,熔点129~131℃(文献值[15]130~131℃)。1H NMR (400MHz,CDCl3)δ:7.08(d,J=10.1Hz,1H),6.26 (dd,J=10.1、1.9 Hz,1H),6.23(dd,J=10.1、1.8 Hz,1H),6.04(dd,J=9.9、1.8 Hz,1H),6.01(s,1H),3.95~3.81(m,4H),2.27(t,J=10.7Hz,1H),2.04(dd,J=15.5、7.2 Hz,1H),1.88~1.79(m,3H),1.70~1.58(m,3H),1.52~1.40(m,3H),1.20(s,3H),0.96(s,3H);13C NMR (101MHz,CDCl3)δ:186.39,162.59,152.95,137.58,128.11,127.73,123.80,118.60,65.31,64.60,48.13,47.61,46.17,41.22,38.33,34.04,30.22,22.14,21.31,20.77,14.29;ESI-MS m/z:327.5 [M+H]+。
向反应瓶中加入4.90g(15mmol)上述合成的化合物8、2.75mL 6mol/L氢氧化钠溶液和100mL甲醇,搅拌溶解,冰浴降温至0℃,滴加7.5mL 30%双氧水,滴毕后撤掉冰浴,室温反应16h,TLC监测反应完全。反应液经减压浓缩至20mL,再加入30mL的水,静置后抽滤出固体,水洗至中性,干燥得到类白色固体4g,收率78%,熔点180~182℃(文献值[15]180~183℃),[α]D:166.2° (c=0.5,dioxane,文献值[9][α]D:165.9°)。1H NMR (400MHz,CDCl3)δ:6.11 (dd,J=9.8、2.3 Hz,1H),6.06(d,J=10.6Hz,1H),5.65(s,1H),3.96~3.84(m,4H),3.60(d,J=4.0Hz,1H),3.47~3.42(m,1H),2.24(t,J=10.6Hz,1H),2.09~2.02(m,1H),2.00~1.93 (m,1H),1.91~1.81(m,2H),1.76~1.62(m,4H),1.56~1.40(m,2H),1.19(s,3H),0.96(s,3H),0.88(t,J=6.7Hz,1H);13C NMR (101MHz,CDCl3)δ:194.81,158.76,139.46,127.94,119.55,118.65,65.34,64.65,59.44,54.69,47.30,46.18,45.86,38.87,37.65,34.02,30.08,22.17,20.66,18.53,14.16;ESI-MS m/z:343.5 [M+H]+。
参考文献[16]方法进行不完全氢化。向200mL的反应瓶中加入2.77g(8mmol)上述合成的化合物9、300mg林德拉催化剂(5%Pd-BaSO4-喹啉)和100mL无水乙醇,室温下氢气催化氢化反应6h。反应结束后抽滤除去固体不溶物,有机层蒸干后残留物用硅胶柱色谱分离,石油醚/丙酮(体积比1:1)洗脱,得到类白色固体粉末1.5g,收率54%,熔点197~199℃,[α]D:82°(c=0.5,dioxane)。1H NMR (400MHz,CDCl3)δ:5.53(t,J=2.1Hz,1H),3.97~3.83 (m,4H),3.64~3.53(m,1H),2.72~2.53(m,2H),2.22~2.03(m,2H),1.93~1.75(m,4H),1.72~1.51(m,4H),1.47~1.33(m,2H),1.32~1.24(m,2H),1.21(s,3H),1.12~1.02(m,2H),0.87(s,3H);13C NMR (101MHz,CDCl3)δ:209.08,128.81,118.40,114.82,77.33,64.22,63.52,49.19,44.88,44.53,41.42,39.51,38.97,35.07,32.84,29.72,24.51,23.56,21.80,17.92,13.39;HRMS(ESI) m/z:C21H31O4[M+H]+,计算值347.2132,实测值347.2125。
向50mL反应瓶中加入0.7g(2mmol)上述合成的化合物10、0.75g(2mmol)七水氯化铈、20mL甲醇和10mL四氢呋喃,待所有固体溶解后0℃下缓慢分批加入152mg(4mmol)硼氢化钠。反应液室温搅拌4h,再加入10mL丙酮猝灭反应,静置后抽滤,除去固体不溶物。有机相减压蒸干后,残留物用环己烷结晶得到类白色针状结晶0.6g,收率86%,熔点197~199℃(文献值[9]195~197℃),[α]D:-87.7° (c=0.5,dioxane,文献值[9][α]D:-87.9°)。1H NMR (400MHz,CDCl3)δ:5.55(t,J=1.8Hz,1H),3.95~3.89 (m,2H),3.89~3.84(m,2H),3.61~3.53(m,1H),3.45(dd,J=11.6、4.4 Hz,1H),2.28~2.18(m,3H),2.07~1.95(m,3H),1.83~1.74(m,1H),1.72~1.64(m,1H),1.60~1.49(m,6H),1.46~1.37(m,3H),1.30~1.16(m,2H),1.04(s,3H),0.87(s,3H);13C NMR(101MHz,CDCl3)δ:138.06,125.43,119.58,68.03,65.19,64.60,63.71,50.51,45.36,42.71,42.39,41.93,35.72,34.08,33.15,31.09,30.88,23.39,22.93,14.24,13.00;ESI-MS m/z:349.5[M+H]+。
向50mL反应瓶中加入348mg(1mmol)化合物7、30mg对甲苯磺酸、5mL水和20mL丙酮。反应液40℃下搅拌12h,减压浓缩至干。残留物用饱和碳酸氢钠溶液30mL和乙酸乙酯(20mL×3)萃取,有机层用水洗涤至中性,无水硫酸镁干燥、过滤、减压浓缩至干。粗产物用甲醇重结晶,得到类白色固体298mg,收率95%,熔点270~272℃(文献值[9]271~273℃),[α]D:+30.5° (c=0.5,MeOH,文献值[9] [α]D:+30.7°)。1H NMR (400MHz,DMSO-d6) δ:5.47(d,J=5.2Hz,1H),4.60(d,J=4.7Hz,1H),4.52(d,J=5.7Hz,1H),3.30~3.20(m,1H),3.20~3.10(m,1H),2.48~2.32(m,2H),2.14~1.92(m,4H),1.89~1.73(m,2H),1.65~1.33 (m,6H),1.28~1.01(m,3H),0.93(s,3H),0.79(s,3H);13C NMR(101MHz,DMSO-d6)δ:220.45,140.12,123.63,77.04,67.09,51.38,50.75,46.95,42.98,42.75,42.42,35.64,32.54,31.96,30.51,23.02,22.05,13.75,13.61;ESI-MS m/z:345.5 [M+H]+。
本研究中1α-羟基去氢表雄酮的合成过程如图式 1所示。以1, 4-雄烯二酮(2)为原料,在酸性条件下用乙二醇保护17位的羰基形成缩酮中间体3,收率97%。文献报道中间体8的合成由17位缩酮保护的去氢表雄酮通过DDQ[4]或Pd/C[9]氧化而得到,通过中间体3的溴代、消除合成化合物8可以有效避免复杂的后处理过程,提高收率,降低成本。中间体9多用液氨-锂体系进行还原,反应条件苛刻、收率不高;参考文献[16]报道的方法,改用活性较低的林德拉催化剂进行不完全还原,同时还原共轭双键和环氧环,得到中间体10,收率55%。氢化反应收率较低,部分原料被完全还原得到完全氢化产物。化合物10再经氯化铈-硼氢化钠-甲醇体系[17]选择性还原3位羰基得到3β-羟基中间体7。氯化铈作为路易斯酸通过配位,加快反应进程,收率86%。最后,在酸性条件下脱去乙二醇保护得到1,收率95%。该路线经6步反应,收率共计29%得到1α-羟基去氢表雄酮。
本文报道了一个简单、有效的1α-羟基去氢表雄酮的合成方法。该方法由1, 4-雄烯二酮出发经过6步反应,以29%的收率得到了1α-羟基去氢表雄酮。该合成路线无需低温及无水无氧等苛刻的反应条件,高效经济,技术可行,安全环保,较适于生产。
H Shimizu, K Shimizu, N Kuboder et al. Org. Proc. Res. Dev., 2005, 9(2): 278~287.
A Perez-Medrano, P A Grieco. J. Am. Chem. Soc., 1991, 113(3): 1057~1059. doi: 10.1021/ja00003a058
Y Matsuya, Y Yamakawa, C Tohda et al. Org. Lett., 2009, 11(13): 3970~3973. https://www.ncbi.nlm.nih.gov/pubmed/19653648
S El Sheikh, A M zu Greffen, J Lex et al. Synlett, 2007, (12): 1881~1884.
C Liu, G D Zhao, X L Mao et al. Steroids, 2014, 85: 569~575. https://www.ncbi.nlm.nih.gov/pubmed/25127149
刘文峥, 杨明波, 孟洪光等.化学试剂, 2014, 36(4): 320~324. http://www.cnki.com.cn/Article/CJFDTotal-HXSJ201404011.htm
C Kaneko, S Yamada, A Sugimoto et al. Tetrahed. Lett., 1973, 14(26): 2339~2342. doi: 10.1016/S0040-4039(01)96213-6
N Hiroyuki, T Sadao, O Kiyoshige et al. JP: 5071456.
Y Z Yin, C Liu, L Q Tang et al. Steroids, 2012, 77(13): 1419~1422. doi: 10.1016/j.steroids.2012.08.018
陈旺, 胡代花, 冯自立等.化学通报, 2018, 81(1): 88~91. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20170705001&flag=1
R M Dodson, A H Goldkamp, R D Muir. J. Am. Chem. Soc., 1960, 82(15): 4026~4033. doi: 10.1021/ja01500a054
A Fujiwara, C Miyamoto, T Okuda. USP: 4379842.
刘佩卉, 刘新利, 鞠培殿等.食品与药品, 2010, 12(7): 256~261. http://www.cnki.com.cn/Article/CJFDTotal-SDPK201007011.htm
M J Gentles, J B Moss, H L Herzog et al. J. Am. Chem. Soc., 1958, 80(14): 3702~3705. doi: 10.1021/ja01547a058
C Kaneko, A Sugimoto, S Yamada et al. Chem. Pharm. Bull., 1974, 22(9): 2101~2107. doi: 10.1248/cpb.22.2101
B Pelc, J Hodková. Collect Czech. Chem. Commun., 1967, 32(1): 410~418. doi: 10.1135/cccc19670410
P Marwah, J B Thoden, D R Powell et al. Steroids, 1996, 61(8): 453~460. doi: 10.1016/0039-128X(96)00092-X

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
