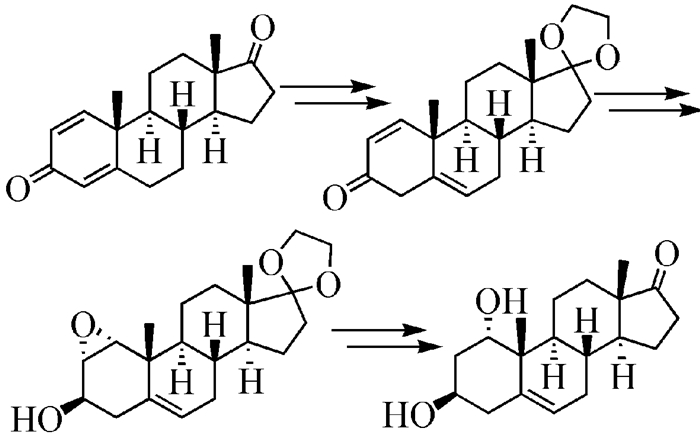
 图式 1
化合物1的合成路线
Scheme1.
Synthetic route for compound 1 in this article
图式 1
化合物1的合成路线
Scheme1.
Synthetic route for compound 1 in this article
引用本文:
陈旺, 胡代花, 冯自立, 王永吉. 1α-羟基去氢表雄酮的合成[J]. 化学通报,
2018, 81(1): 88-91.

Citation: Chen Wang, Hu Daihua, Feng Zili, Wang Yongji. Synthesis of 1α-Hydroxydehydroepiandrosterone[J]. Chemistry, 2018, 81(1): 88-91.
Citation: Chen Wang, Hu Daihua, Feng Zili, Wang Yongji. Synthesis of 1α-Hydroxydehydroepiandrosterone[J]. Chemistry, 2018, 81(1): 88-91.
1α-羟基去氢表雄酮的合成
摘要:
本文以1,4-雄烯二酮为起始原料,经羰基保护、溴代、异构化、羰基还原、环氧化、开环及脱保护7步反应以28.3%的总收率合成1α-羟基去氢表雄酮。该方法具有高效经济、技术可行、安全环保等优点,为1α-羟基去氢表雄酮的规模生产及维生素D类药物合成研究奠定理论基础。
-
关键词:
- 1α-羟基去氢表雄酮
- / 1, 4-雄烯二酮
- / 环氧化
- / 合成
English
Synthesis of 1α-Hydroxydehydroepiandrosterone
Abstract:
1α-hydroxydehydroepiandrosterone was prepared from androsta-1, 4-diene-3, 17-dione through seven steps such as protection of carbonyl group, bromination, deconjugation, reduction, epoxidation, ring-opening and deprotection, with an overall yield of 28.3%. The method has the advantages of high efficiency and economy, technical feasibility, safety and environmentally benign, and lays a foundation for industrialized production of 1α-hydroxydehydroepiandrosterone and research of vitamin D drugs.
-
1α-羟基去氢表雄酮属于甾体类化合物,是合成马沙骨化醇、他卡西醇、阿法骨化醇、氟骨三醇等活性维生素D3类药物的关键中间体[1],也可用于合成其他天然产物如(+)-醉茄内酯(Withanolide) E[2]、Sominone及其衍生物[3]、Cyclocitrinols[4]等,同时也是合成维生素D类似物的重要原料[5]。但由于1α-羟基去氢表雄酮原料匮乏,其合成条件苛刻、收率较低,一定程度制约了活性维生素D类药物的研发。因此,开发高效经济、技术可行、安全环保、适于工业化生产的1α-羟基去氢表雄酮合成工艺具有重要现实意义和广阔应用前景。
目前,文献报道的1α-羟基去氢表雄酮(化合物1,结构见图式 1)合成方法主要为化学合成法和生物转化法[6]。1973年,Kaneko等[7]以1, 4-雄烯二酮为原料,经羰基保护、碱催化的烯键异构、Ca(BH4)2还原、硼氢化氧化制得lα, 3β-二羟基(中间体7,15%)和2α, 3β-二羟基(20%)两种构型产物;1α羟基产物再在酸性条件下脱去保护基,最终制得1,整条路线收率小于7%。该路线由于收率低且使用毒性较大的硼烷,故不适用于工业化生产。Hiroyuki等[8]将中间体5经三氟乙酸汞处理和硼氢化钠还原得中间体7,其收率为36.5%,但该路线使用了三氟乙酸汞,也不适于大生产。2007年,Sheikh等[4]在合成天然化合物Cyclocitrinols的过程中合成了乙酰基保护的1。以去氢表雄酮为原料,经羰基保护、DDQ氧化脱氢得到3-酮-1, 4, 6-三烯-雄甾-17, 17-乙二醇缩酮、再经碱性双氧水环氧化、液氨电子还原反应得到中间体7,化合物7脱去保护基得到1,总收率为21%。虽然Sheikh等合成方法较Kaneko等的方法在收率上有很大提高,但该路线因涉及DDQ脱氢氧化反应处理困难,仍不适于工业化生产。针对以上问题,刘兆鹏等[9]对该工艺路线进行改进。在烯丙基磷酸二乙酯存在下利用钯碳将乙二醇保护的去氢表雄酮脱氢得到中间体3-酮-1, 4, 6-三烯-雄甾-17, 17-乙二醇缩酮,但该方法使用烯丙基磷酸二乙酯的量较大,生产成本较高,且锂氨还原需在-40℃到-80℃的低温下进行,条件较苛刻。
图式 1
化合物1的合成路线
Scheme1.
Synthetic route for compound 1 in this article
微生物转化法合成化合物1的探索已经进行了半个多世纪。Dodson等[10]采用Penicillium.sp ATCC 12556菌种通过微生物发酵合成化合物1,粗品收率约33.3%,但生产成本较高。Fujiwara等[11]采用菌种Penicillium Oxalicum IFO-7000合成了化合物1,收率为45.0%。刘佩卉等[12]用斜卧青霉(Penicillium decumbens)UC086合成了化合物1,收率为33.4%。
微生物转化法生产周期长、生产成本高,需要提取与柱层析等复杂的过程,使得该类方法的应用受到限制。对于大规模生产而言,生物合成法还需进行深入研究以适应工业化生产的要求。因此,探索一条原料易得、操作简便、条件温和、适于工业化生产的合成路线具有重要现实意义。本文报道以1, 4-雄烯二酮为原料,经羰基保护、溴代、异构化、还原、环氧化、开环和水解7步反应以28.3%总收率合成化合物1(合成路线见图式 1),该合成路线高效经济、技术可行、安全环保、较适于工业化生产。
1 实验部分
1.1 仪器和试剂
X4型显微熔点仪(温度未校正);Avance-400型核磁共振谱仪(Brucker公司),TMS为内标;LTQ-Orbitrap XL型质谱仪(Thermo Fisher公司),离子源采用电喷雾离子化(ESI);BWXG4型圆盘旋光仪。柱色谱使用200~300目硅胶(青岛海洋化工厂),薄层色谱使用Silica Gel GF254(青岛海洋化工厂);所有试剂均为市售分析纯级试剂,无水溶剂用常规方法干燥处理。
1.2 化合物1的合成
1.2.1 3-酮-1, 4-二烯-雄甾-17, 17-乙二醇缩酮(3)的合成
向安装有分水器的250mL反应瓶中加入7.11g (25mmol)1, 4-雄烯二酮、3.1g(50mmol)乙二醇、100mg对甲苯磺酸和100mL苯,搅拌加热、回流反应18h,TLC检测反应完全。反应液降至室温,用饱和碳酸氢钠水溶液100mL和乙酸乙酯(50mL×3)萃取,合并有机层,无水硫酸镁干燥,减压浓缩。乙酸乙酯结晶,得到类白色固体粉末7.7g,收率94%,熔点170~171℃(文献值[13]171~172℃)。1H NMR(400MHz,CDCl3)δ:7.06 (d,J=10.1Hz,1H),6.23(dd,J=10.1、1.9 Hz,1H),6.07(t,J=1.5Hz,1H),3.93~3.80(m,4H),2.52~2.41(m,1H),2.40~2.32(m,1H),2.05~1.93(m,2H),1.85~1.74(m,2H),1.73~1.53(m,4H),1.48~1.26(m,3H),1.23(s,3H),1.14~1.00(m,2H),0.92(s,3H);13C NMR(101MHz,CDCl3)δ:186.37,169.15,155.84,127.48,123.86,118.93,65.24,64.55,52.16,49.30,45.94,43.58,35.81,34.03,32.91,32.79,30.24,22.78,22.30,18.73,14.37;ESI-MS m/z:329.5[M+H]+。
1.2.2 3-酮-1, 5-二烯-雄甾-17, 17-乙二醇缩酮(4)的合成
向250mL反应瓶中加入3.28g (10mmol)上述合成的化合物3、2.67g (15mmol)N-溴代丁二酰亚胺、100mg偶氮二异丁腈和100mL四氯化碳,搅拌加热、回流反应6h,TLC检测反应完全。反应液降至室温,抽滤除去固体不溶物,滤饼用四氯化碳洗涤,滤液减压浓缩至干。该溴代产物不经纯化直接使用。向残留物中加入9.75g (150mmol)锌粉、100mL乙醇和25mL水,50℃下搅拌反应22h,TLC检测反应完全。反应液抽滤除去固体,滤液减压浓缩至干。粗产物经硅胶柱色谱纯化,石油醚/丙酮(体积比10:1)洗脱,得到类白色固体粉末2.46g,两步收率共计75%,熔点156~158℃(文献值[14]154~157℃)。1H NMR (400MHz,CDCl3)δ:7.06(d,J=10.1Hz,1H),6.23(d,J=10.1,1H),6.07(t,J=1.5Hz,1H),3.93~3.80(m,4H),2.52~2.32(m,2H),2.05~1.93(m,2H),1.84~1.73(m,2H),1.72~1.57(m,4H),1.48~1.26(m,3H),1.23(s,3H),1.15~1.01(m,2H),0.92(s,3H);13C NMR(101MHz,CDCl3)δ:186.37,169.12,155.82,127.48,123.87,118.93,65.24,64.55,52.16,49.30,45.94,43.57,35.81,34.03,32.91,32.79,30.24,22.78,22.30,18.73,14.37;ESI-MS m/z:329.5 [M+H]+。
1.2.3 3β-羟基-1, 5-二烯-雄甾-17, 17-乙二醇缩酮(5)的合成
向100mL反应瓶中加入1.31g (4mmol)上述合成的化合物4、1.49g (4mmol)七水氯化铈、20mL甲醇和10mL四氢呋喃,待所有固体溶解后0℃下缓慢分批加入304mg (8mmol)硼氢化钠。反应液室温搅拌4h,再加入丙酮10mL猝灭反应,静置后抽滤,除去固体不溶物。有机相减压蒸干后残留物用环己烷重结晶得到类白色针状结晶1.22g,收率92%,熔点136~138℃(文献值[14]134~138℃)。1H NMR (400MHz,CDCl3)δ:5.78(dd,J=10.1、1.8 Hz,1H),5.54(dt,J=10.1、1.4 Hz,1H),5.43~5.39(m,1H),4.25~4.17(m,1H),3.95~3.89(m,2H),3.86~3.81(m,2H),2.49~2.44(m,1H),2.33~2.25(m,1H),2.09~1.96(m,2H),1.85~1.65(m,4H),1.65~1.49(m,4H),1.48~1.42(m,2H),1.31~1.24(m,1H),1.13~1.04(m,4H),0.88(s,3H);13C NMR(101MHz,CDCl3)δ:138.88,136.26,129.43,121.59,119.39,69.72,65.19,64.57,50.67,46.59,45.76,40.44,38.51,34.20,32.06,30.66,30.51,22.70,21.90,20.46,14.29;ESI-MS m/z:331.5 [M+H]+。
1.2.4 1α, 2α-环氧-5-烯-3β-羟基-雄甾-17, 17-乙二醇缩酮(6)的合成
向100mL反应瓶中加入3mol/L过氧化叔丁醇甲苯溶液(2.5mL,7.5mmol),-10℃下缓慢滴加入2.13g(7.5mmol)钛酸四异丙酯,待溶液呈深棕色后缓慢滴加入1.04g(3mmol)上述合成的化合物5的10mL氯仿溶液,室温下搅拌3h,再加入50mL氯仿和10mL水。反应液抽滤除去固体不溶物,有机层减压蒸干,残留物经硅胶柱色谱纯化,石油醚/丙酮(体积比5:1)洗脱,得到类白色固体粉末572mg,收率55%;同时得到246mg化合物4(25%),并回收原料187mg(18%),熔点162~163℃。1H NMR(400MHz,CDCl3)δ:5.32(t,J=4.4Hz,1H),3.98~3.92(m,2H),3.92~3.85(m,3H),3.30(d,J=3.9Hz,1H),3.27(d,J=4.0Hz,1H),2.21~2.10(m,2H),2.09~1.99(m,2H),1.88~1.80(m,1H),1.80~1.75(m,1H),1.73~1.67(m,2H),1.67~1.56(m,4H),1.53~1.44(m,2H),1.35~1.26(m,1H),1.23~1.16(m,1H),1.13(s,3H),0.92(s,3H);13C NMR(101MHz,CDCl3)δ:138.24,121.68,119.28,70.91,65.23,64.61,61.41,57.79,50.73,45.94,44.69,36.15,35.20,34.24,31.85,30.60,30.28,22.62,21.16,17.44,14.38;HRMS (ESI):C21H31O4 [M+H]+,计算值347.2132,实测值347.2127。
1.2.5 1α, 3β-二羟基-5-烯-雄甾-17, 17-乙二醇缩酮(7)的合成
向50mL反应瓶中加入450mg(1.3mmol)上述合成的化合物6和20mL四氢呋喃,室温下分批加入61mg(1.6mmol)四氢铝锂,加毕回流反应4h。反应结束后降至室温,加入1mL水,过滤、有机层减压浓缩至干,残留物用乙酸乙酯和石油醚结晶得到类白色针状结晶385mg,收率85%,熔点197~199℃(文献值[9]195~197℃),αD:-87.7° (c=0.5,dioxane,文献值[9] αD:-87.9°)。1H NMR(400MHz,CDCl3)δ:5.55(t,J=1.8Hz,1H),3.95~3.89(m,2H),3.89~3.84(m,2H),3.61~3.53(m,1H),3.45(dd,J=11.6,4.4Hz,1H),2.28~2.18(m,3H),2.07~1.95(m,3H),1.83~1.74(m,1H),1.72~1.64(m,1H),1.60~1.49(m,6H),1.46~1.37(m,3H),1.30~1.16(m,2H),1.04(s,3H),0.87(s,3H);13C NMR(101MHz,CDCl3)δ:138.06,125.43,119.58,68.03,65.19,64.60,63.71,50.51,45.36,42.71,42.39,41.93,35.72,34.08,33.15,31.09,30.88,23.39,22.93,14.24,13.00;ESI-MS m/z:349.5[M+H]+。
1.2.6 化合物1的合成
向50mL反应瓶中加入348mg(1mmol)上述合成的化合物7、30mg对甲苯磺酸、5mL水和20mL丙酮。反应液40℃下搅拌12h,减压浓缩至干;残留物用30mL饱和碳酸氢钠溶液和乙酸乙酯(20mL×3)萃取,有机层用水洗涤至中性,无水硫酸镁干燥、过滤、减压浓缩至干;甲醇结晶,得到类白色固体298mg,收率95%,熔点270~272℃(文献值[9]271~273℃),αD:+30.5° (c=0.5,MeOH,文献值[9] αD:+30.7°)。1H NMR(400MHz,DMSO-d6)δ:5.47 (d,J=5.2Hz,1H),4.60(d,J=4.7Hz,1H),4.52(d,J=5.7Hz,1H),3.30~3.20(m,1H),3.20~3.10(m,1H),2.48~2.32(m,2H),2.14~1.92(m,4H),1.89~1.73(m,2H),1.65~1.33(m,6H),1.28~1.01(m,3H),0.93(s,3H),0.79(s,3H);13C NMR(101MHz,DMSO-d6)δ:220.45,140.12,123.63,77.04,67.09,51.38,50.75,46.95,42.98,42.75,42.42,35.64,32.54,31.96,30.51,23.02,22.05,13.75,13.61;ESI-MS m/z:345.5 [M+H]+。
2 结果与讨论
本文中化合物1的合成过程如图式 1所示。以1, 4-雄烯二酮(2)为原料,在酸性条件下用乙二醇保护17位的羰基形成缩酮中间体3,收率94%。文献报道[7]化合物3在DMSO为溶剂、叔丁醇钾为碱的作用下即可去共轭异构化为化合物4,然而采用文献报道的方法不能按照预期的高收率得到化合物4。因此,化合物3经烯丙位溴代、锌粉还原得到中间体4,两步收率共计75%。化合物4再经氯化铈-硼氢化钠-甲醇体系[15]选择性还原3位羰基得到3β-羟基中间体5,收率92%。化合物4对碱性条件极其敏感,在极微弱碱和质子溶剂的作用下就异构化为化合物3,因此不能用双氧水在碱性条件下氧化化合物4然后再还原直接得到化合物7。化合物5再经钛(Ⅳ)催化的环氧化反应[16]选择性地引入lα, 2α-环氧环,得到环氧中间体6,收率55%;同时得到羟基氧化的副产物化合物4(25%)及回收部分原料(18%)。该环氧化过程参考文献报道,由Sharpless氧化改造而得,若在体系中加入L-(+)-酒石酸二乙酯则主要得到1β, 2β-环氧产物。若在体系中加入D-(-)-酒石酸二乙酯则由于3位立体构型的限制几乎不反应。该环氧中间体12经四氢锂铝还原环氧环,选择性地得到1α-羟基中间体7,收率85%。最后,在酸性条件下脱去乙二醇保护得到1,收率95%。该路线经7步反应,以28.3%的总收率得到化合物1。
3 结论
本文报道了一个新的简单、有效的1α-羟基去氢表雄酮的合成方法。该方法由1, 4-雄烯二酮出发经过7步反应,以28.3%的收率得到了1α-羟基去氢表雄酮。该合成路线无低温、无水无氧反应,高效经济、技术可行、安全环保、较适于工业化生产。
-
-
[1]
H Shimizu, K Shimizu, N Kuboder et al. Org. Process. Res. Dev., 2005, 9(2):278~287.
-
[2]
A Perez-Medrano, P A Grieco. J. Am. Chem. Soc., 1991, 113(3):1057~1059. doi: 10.1021/ja00003a058
-
[3]
Y Matsuya, Y Yamakawa, C Tohda et al. Org. Lett., 2009, 11(13):3970~3973. doi: 10.1021/ol901553w
-
[4]
S El Sheikh, A M zu Greffen, J Lex et al. Synlett, 2007, 2007(12):1881~1884. doi: 10.1055/s-2007-984521
-
[5]
C Liu, G D Zhao, X L Mao et al. Steroids, 2014, 85:569~575.
-
[6]
刘文峥, 杨明波, 孟洪光等.化学试剂, 2014, 36(4):320~324. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxsj201404011&dbname=CJFD&dbcode=CJFQ
-
[7]
C Kaneko, S Yamada, A Sugimoto et al. Tetrahed. Lett., 1973, 14(26):2339~2342. doi: 10.1016/S0040-4039(01)96213-6
-
[8]
N Hiroyuki, T Sadao, O Kiyoshige et al. JP:5071456.
-
[9]
Y Z Yin, C Liu, L Q Tang et al. Steroids, 2012, 77(13):1419~1422. doi: 10.1016/j.steroids.2012.08.018
-
[10]
R M Dodson, A H Goldkamp, R D Muir. J. Am. Chem. Soc., 1960, 82(15):4026~4033. doi: 10.1021/ja01500a054
-
[11]
A Fujiwara, C Miyamoto, T Okuda. USP:4379842.
-
[12]
刘佩卉, 刘新利, 鞠培殿等.食品与药品, 2010, 12(7):256~261. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=sdpk201007011&dbname=CJFD&dbcode=CJFQ
-
[13]
M J Gentles, J B Moss, H L Herzog et al. J. Am. Chem. Soc., 1958, 80(14):3702~3705. doi: 10.1021/ja01547a058
-
[14]
C Kaneko, A Sugimoto, S Yamada et al. Chem. Pharm. Bull., 1974, 22(9):2101~2107. doi: 10.1248/cpb.22.2101
-
[15]
P Marwah, J B Thoden, D R Powell et al. Steroids, 1996, 61(8):453~460. doi: 10.1016/0039-128X(96)00092-X
-
[16]
D Scheddin, H Mayer, S Wittmann et al. Steroids, 1996, 61(10):598~608. doi: 10.1016/S0039-128X(96)00120-1
-
[1]
-


 下载:
下载:
 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
