 图1
氧化石墨烯的功能化常用方法[13]
Figure1.
Common methods for functionalization of graphene oxide[13]
图1
氧化石墨烯的功能化常用方法[13]
Figure1.
Common methods for functionalization of graphene oxide[13]
引用本文:
雷颖, 杨蓉, 王黎晴, 李兰, 杨文宇, 苏香香, 路蕾蕾. 石墨烯的功能化及其在储能材料领域中的应用[J]. 化学通报,
2017, 80(9): 802-808.

Citation: Lei Ying, Yang Rong, Wang Liqing, Li Lan, Yang Wenyu, Su Xiangxiang, Lu Leilei. The Functionalization of Graphene and Their Applications in the Field of Energy Storage Materials[J]. Chemistry, 2017, 80(9): 802-808.
Citation: Lei Ying, Yang Rong, Wang Liqing, Li Lan, Yang Wenyu, Su Xiangxiang, Lu Leilei. The Functionalization of Graphene and Their Applications in the Field of Energy Storage Materials[J]. Chemistry, 2017, 80(9): 802-808.
石墨烯的功能化及其在储能材料领域中的应用
摘要:
石墨烯是由sp2杂化的碳原子紧密堆积成的单原子层二维碳材料,由于其优异的物理和化学性质被视为最有前景的新型材料之一。但由于石墨烯片层之间在范德华力的作用下易发生不可逆团聚,丧失其单层二维纳米片的结构特性,以及石墨烯表面呈现惰性状态,致使其与其他介质的相互作用较弱,难以均匀分散在极性或非极性的溶剂中,因而石墨烯的应用受到限制。对石墨烯进行功能化可以调控其分子结构、电子能级和化学性质,不仅可以有效抑制石墨烯的团聚而且能够改善其在溶剂中的分散性和稳定性,从而实现石墨烯基材料的多元化应用。本文综述了近年来共价键和非共价键功能化石墨烯以及其复合材料在储能领域的研究进展,并对功能化石墨烯的发展前景进行了展望。
English
The Functionalization of Graphene and Their Applications in the Field of Energy Storage Materials
Abstract:
Graphene is a two-dimensional material that is compactly packed with sp2 hybridized carbon atoms and is considered as one of the most promising new materials due to its excellent physical and chemical properties. However, because of the irreversible agglomeration tendency of the graphene sheets, the structural properties of the monolayer nanosheets are disappeared easily. Moreover, the surface of the graphene is in an inert state, resulting in weak interaction with other media, making it difficult to disperse uniformly in a polar or non-polar solvent, so the application of graphene is limited. The functionalization of graphene can control its molecular structure, electronic energy level and chemical properties, which can not only inhibit the agglomeration of graphene effectively, but also improve its dispersibility and stability in the solvent, achieving the diversification of graphene-based materials. In this paper, the progress in functionalized of graphene and its composites in application as energy storage materials are reviewed, and the development prospect of functionalized graphene is prospected as well.
-
Key words:
- Graphene
- / Functionalization
- / Materials For Energy Storage
-
石墨烯是一种由单层碳原子紧密堆积成二维蜂窝状晶格结构的新型材料[1],自2004年被发现以来,就成为科学研究的焦点。这种二维纳米材料的基本结构单元是有机材料中最稳定的苯六元环,仅为一个碳原子的厚度(0.335nm),是迄今为止发现的最薄的二维材料。这种特殊的结构使得石墨烯呈现出优异的物化性质[2]。石墨烯的理论比表面积高达2600m2/g[3],其结构中长程有序的π键电子结构使石墨烯具有优异的导热性能(3×103W/(m·K))、力学性能(1.06×103GPa)和室温下高的电子迁移率(1.5×104cm2/(V·s))[4]。但是要充分发挥其优势,仍需克服较多困难。例如,石墨烯的带隙为零[5],限制了其在半导体行业和电化学传感器中的应用;石墨烯结构规整,与有机溶剂作用力弱,很难分散在其中;石墨烯层与层之间很容易在范德华力的作用下发生团聚,因而其应用受到限制[6, 7]。
为了解决上述问题,满足应用的需求,许多学者在石墨烯的基础上添加其他成分和结构形成一类新材料——功能化石墨烯[8],它们在保持石墨烯大部分基本特性的同时,具有不同于本征石墨烯的新性能[9]。由于功能化基团的引入,功能化石墨烯相对于石墨烯而言,不仅在溶剂中的分散性和溶解力显著增加,而且充分发挥了石墨烯在力学、光学、电学及热学上的优异性能。同时,根据不同修饰方法可以合理设计针对实际需求的功能化石墨烯,目前功能化采用的主要方法有共价键功能化和非共价键功能化。共价键功能化是利用氧化石墨烯(GO)上的反应性基团(环氧基、羟基、羧基)与小分子或聚合物反应接枝,而非共价键功能化是利用改性剂与石墨烯之间π-π键、离子键、氢键的相互作用。本文将重点介绍近年共价键功能化石墨烯、非共价键功能化石墨烯以及石墨烯基功能化复合材料在储能领域中的研究进展,并对其今后的研究方向作了展望。
1 石墨烯的共价键功能化
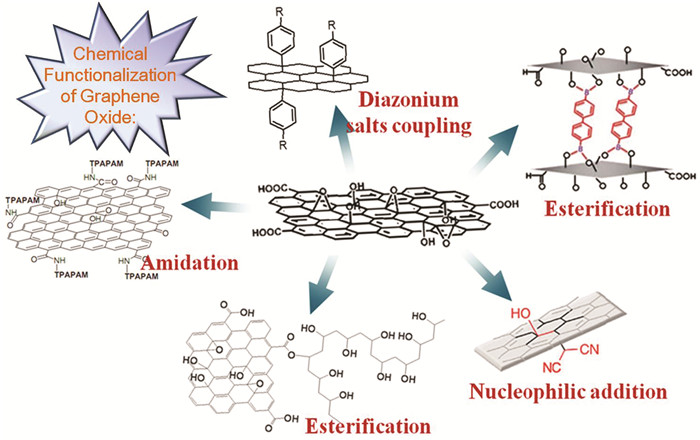
石墨烯的共价键功能化[10, 11]是基于共价键对石墨烯的边沿或缺陷处进行化学修饰,通过共价键功能化可以提高石墨烯的液相可加工性,制备的石墨烯材料比较稳定,并能够在最大程度上保持石墨烯本征属性的同时赋予其新的性能[12]。但是完美的石墨烯呈现化学惰性,非常稳定,因此通常利用化学性质较为活泼的GO作前驱体与功能分子相作用,制备出具有特殊性质的功能化石墨烯。GO表面存在着大量的羧基、羟基、环氧基等含氧官能团,兼有芳香结构的碳骨架,因此对GO的共价键功能化主要围绕这些活性位点展开,常见共价键功能化路线如图 1所示[13]。
1.1 有机小分子共价键功能化
通过有机小分子对石墨烯进行功能化,使小分子中的一部分基团通过共价键连接在石墨烯上,从而可以改善石墨烯在溶剂中的分散性和稳定性[14]。目前,石墨烯的有机小分子共价键功能化主要依靠GO表面羟基、环氧基的亲核开环反应、异氰酸酯化反应、共轭平面的重氮化、环加成反应和硅烷化反应等进行。
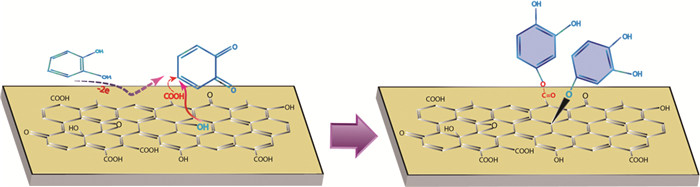
Jokar等[15]利用电合成过程将邻苯二酚经过氧化作用转换为1, 2-苯醌,与部分还原的GO表面的羟基连接(如图 2所示),进行共价键合得到功能化石墨烯,用作高性能超级电容器电极材料。该电极在28A/g的电流密度下比容量达288F/g,表现出良好的倍率性能和优异的循环稳定性,显著地提高了超级电容器的性能。并且,电解液pH的变化对该电极的氧化还原行为有显著影响,在pH为3~7范围内,随着pH增加,阴极峰转向更负的电位。
艾伟[13]利用多聚磷酸催化的环化过程,通过GO片层上的羧基与邻羟基苯胺上的羟基及邻苯二胺上的氨基进行反应,制备了两种功能化GO(BO-G和BI-G)。研究发现,功能化后的GO在有机溶剂中的溶解度增加,在进一步还原过程中,功能化基团不但在一定程度上阻止石墨烯的聚集,而且还增加了电极与电解液之间的电荷转移过程,用于超级电容器电极表现出高的比电容以及优良的循环稳定性。循环2000次后,BO-G的比电容值没有发生大的变化,且在最初的500次循环中,比电容值在不断地增加;BI-G在2000次循环后,比电容保持率为85%。
近年来对提升锂离子电池性能的研究不断取得进展,但仍有些问题制约着人们获得关键性突破。例如,Li+迁移过慢、电极的电子传导性差、高充放电率下电极与电解液间的电阻率增大等现象[12]。为了提高锂电池的充放电效率,人们将石墨烯引入到电极材料中。杜祝祝[16]通过酯化反应将4-羟基-2, 2, 6, 6-四甲基哌啶氧(4-OH-TEMPO)和GO共价键相连,GO在功能化的同时被还原,制备得到了TEMPO-G。得到的TEMPO-G材料包含了高导电的石墨烯框架体系和丰富的电化学活性氮氧自由基。用于锂离子电池的阴极材料时,TEMPO-G表现出较高的比容量和良好的循环稳定性,循环400次后,TEMPO-G的放电比容量保持为1080mAh/g。
GO和修饰试剂之间的化学反应往往缺乏足够的可控性,功能基团的连接数量主要依赖于GO上含氧官能团的种类和数目。此外,功能基团的引入也改变了石墨烯的本征特性,往往造成石墨烯结构与性质的明显改变。因此,迫切需要开发一种有效的GO还原和可控的功能化方法,在保持石墨烯本征特性的基础上改善其不可逆团聚现象。刘跃文等[17]利用金属锂萘四氢呋喃体系产生的溶剂化电子还原GO,很大程度上恢复了石墨烯的导电性(1361S/m);然后通过可控的还原烷基化反应在石墨烯表面引入烷基链,在保证石墨烯本征特性的基础上显著改善了其分散性。由于大量存在的褶皱结构,烷基功能化石墨烯用作锂离子电池的负极活性材料时首次充放电比容量和电池循环性能都明显高于石墨烯本身。
汪丽丽等[18]利用水合肼还原十八胺(ODA)接枝的GO,得到十八胺功能化石墨烯(ODA-G),将ODA-G与聚苯胺(PANI)通过溶液共混法制备了功能化石墨烯的复合材料(ODA-G/PANI)。在电流密度为1A/g时,2(wt)%ODA-G/PANI复合材料的比电容为787F/g,远远高于纯PANI的比电容(426F/g);200次循环后容量保持率为96%。此外,该复合材料还具有优良的功率特性,是理想的超级电容器电极材料。刘赞辉等[19]采用酰胺化法在GO上以共价键接枝聚对苯二胺(PPD),进一步还原去除未反应的含氧官能团后,制备了一种高性能的新型电容器用电极材料rGO/PPD。在1A/g的电流密度下该电极的比电容高达347F/g,远高于纯组分rGO和PPD的比容量;在10A/g电流密度下循环1000次后,容量保持率为90.1%,呈现出优异的长期稳定储电性能。
Song等[20]采用两步水热法通过共价键将胺分子水平接枝到石墨烯表面,XRD显示胺分子充当分子间隔物以扩大石墨烯层间距和比表面积。功能化后的层间距值从0.84nm增加到1.23nm,GO还原之后间隔变化可忽略,这意味着3D石墨烯纳米结构的高稳定性。该石墨烯材料在水性和有机电解质中的电容均提高,表现出快速离子扩散特征。该材料在离子液体电解质中最大比电容达119F/g,并具有超高能量密度(51Wh/kg)以及低的自放电率。他们认为使用脂肪族胺可以更好地阐明间隔效应和电双层电容之间的相关性。Song等[21]还分别用邻苯二胺(OPD)、间苯二胺(MPD)和对苯二胺(PPD)制备了功能化石墨烯(PD/rGO)。研究显示,PPD和MPD分子可以分别将石墨烯层间距增大到1.41和1.30 nm,而OPD分子无序地结合或未结合到石墨烯层的基面,导致小而多变的层间距。电化学研究表明,PPD/rGO的最大比电容为422F/g,并具有优良的循环稳定性以及低电荷转移电阻。
1.2 聚合物共价键功能化
聚合物共价键功能化主要包括两种方法,分别为分散法和收敛法。分散法是引发通过键接在石墨烯表面的单体在石墨烯或GO的表面或者边缘部位发生接枝聚合[14]而实现功能化。Liu等[22]通过对苯二胺重氮加成反应接枝生成氨基官能化石墨烯片(AFG),然后将AFG与苯胺原位化学氧化聚合制成聚苯胺石墨烯复合材料(PAFG)。研究表明,PAFG电极具有较高的比电容(1295F/g,1A/g)。Cha等[23]先制备了聚多巴胺(PDA)官能化GO,再与聚(3, 4-亚乙基)形成PDA-GO/PEDOT复合薄膜。用作超级电容器的电极材料时,在1A/g的电流密度下该电极的比电容约为126F/g,是GO/PEDOT复合膜的两倍。
收敛法是通过聚合物上的官能团与石墨烯或GO上的官能团反应实现的,在石墨烯上引入聚合物引发基团然后原位生长聚合物,这种方法主要依赖于GO表面的特定引发剂[14]。Qiu等[24]利用乙二胺与GO在95℃条件下反应得到功能化氧化石墨烯(frGO),将frGO分散在乙醇的水溶液中,加入过硫酸铵和苯胺的盐酸溶液,发生原位聚合反应,得到frGO/PANI复合材料。用于超级电容器时,该复合材料具有优异的电化学性能,在0.5A/g电流密度下比电容达489F/g,在2A/g的电流密度下经过1000次循环后容量保持率为85%。
Lingappan等[25]首先将GO与亚硫酰氯进行处理,得到酰化的GO,随后将其用于活化2-乙炔基吡啶(2-EPY)的聚合反应得到聚(2-乙炔基吡啶)官能化石墨烯(rGO/P2EP)。由于rGO和P2EP的协同作用,当该复合材料用作超级电容器的电极时,其在0.5A/g电流密度下的比电容为225F/g,并具有优异的倍率性能和长期循环稳定性(在1000次循环后容量保持率为94%)。
通过在石墨烯片的边缘处选择性地接枝化学基团可以赋予石墨烯溶解性、成膜能力和电催化活性,同时在很大程度上保持原始石墨烯的物理化学性质。Xiang等[26]对边缘功能化石墨烯材料(EFG)和2D共价有机聚合物(2D COP)作为能源材料的应用进行了综述。
2 石墨烯的非共价键功能化
石墨烯的非共价键功能化主要是利用功能分子与石墨烯片层之间的超分子作用或范德华力作用,获得具有特定功能的石墨烯基复合材料。这种功能化方法最大的优点就是操作简单、条件温和、对石墨烯的结构破坏非常小,可以最大程度地保留石墨烯的本征特性[8]。但是由于功能分子与石墨烯复合的作用力较小,因此复合材料的稳定性相对较差。对于完整的石墨烯而言,非共价键功能化是通过石墨烯片层上sp2杂化的碳原子与功能分子之间的π-π作用来完成的。对于化学氧化法制备的GO而言,由于存在大量的缺陷和含氧基团,非共价键功能化既可以利用π-π作用,也可以利用离子键或者氢键等来实现。
2.1 π-π键功能化
不论是石墨烯还是GO都含有大量sp2杂化的碳原子,都可以与具有π共轭结构的有机分子通过π-π相互作用来进行功能化。
王欢文[27]通过非共价键修饰,在石墨烯表面引入叔丁基对苯二酚。苯环与石墨烯形成π-π作用,叔丁基的疏水性增强了它在石墨烯片上的附着能力。通过叔丁基对苯二酚对石墨烯纳米片进行修饰后,其循环伏安的背景电流增大,而且在双电层背景电流的基础上又引入了一对氧化还原峰,从而使体系的电容性能明显提高。结果表明,叔丁基对苯二酚修饰的石墨烯纳米片的比电容高达302F/g,其比电容较纯石墨烯提高了51%。同时,修饰后的石墨烯表现出更好的倍率特性,当放电电流密度从0.25A/g增加到8A/g时,叔丁基对苯二酚修饰的石墨烯纳米片的电容保持率达63%,而纯石墨烯电容保持率却只有25%。此外,叔丁基对苯二酚修饰的石墨烯电极具有长的循环寿命,充放电循环800次后,电容保持率仍高达94%。
Han等[28]通过引入使用原位聚合磺酸官能化的石墨烯(SFG)制备了聚(3, 4-乙烯二氧噻吩)(PEDOT)/磺酸官能化的石墨烯复合水凝胶,由于引入了SFG,该复合水凝胶的比电容显著提高,当SFG与3, 4-乙烯二氧噻吩(EDOT)的进料比低至1:1000时,复合水凝胶的比电容可以达到93F/g,比纯PEDOT水凝胶(39F/g)高了2倍。当KI和K3Fe(CN)6-H2SO4溶液分别用作氧化还原活性的电解质时,该复合水凝胶在5A/g的电流密度下的比电容分别为135和220 F/g。
Fan等[29]将对甲氧基锌卟啉-富勒烯衍生物(ZnPp-C60)和电化学还原的氧化石墨烯(ERGO)通过π-π作用非共价结合,得到可用于电催化方面的新型界面材料ERGO@ZnPp-C60。ZnPp-C60的存在使得ERGO具有更高的氧化还原可逆性和电催化活性。
2.2 离子键功能化
GO由于含有大量带负电的含氧官能团(羟基、羧基和环氧基等),片层间的作用力表现为静电斥力,可以稳定分散在水中,所以也可以通过引入带电离子对其进行功能化。例如,Shao等[30]以介孔阳极三氧化二铝(AAO)为模板,利用AAO的含氧官能团与石墨烯表面含氧基团之间的静电作用,使石墨烯在AAO表面发生自组装。
Dong等[31]将GO用NaBH4预还原,采用4-氨基偶氮苯-4’-磺酸盐(SAS)进行重氮化得到其重氮盐(ADS),在碳酸钠的弱碱性溶液中冰浴下ADS与预还原氧化石墨烯进行反应,之后在肼的还原作用下得到表面改性氧化石墨烯(ADS-G)。结果表明,ADS-G具有更好的水分散性和高的比电容(210F/g),电导率可达到1120S/m,有望作为良好的电极材料用于能量存储。
2.3 氢键功能化
氢键是一种较强的非共价键,由于GO的表面具有大量的羧基和羟基等极性基团,容易与其他物质产生氢键相互作用,因此,可以利用氢键对GO进行功能化。石墨烯的氢键功能化不仅可以用于提高石墨烯的溶解性,还能利用氢键实现有机分子在石墨烯上的负载。Yang等[32]利用抗肿瘤药物盐酸阿霉素(DRX)上的羟基和氨基与GO表面的羟基和羧基间的氢键作用,制备了一种新型GO-DXR杂化体,并研究了DXR在GO表面的负载和控释行为。Patil等[33]利用化学法合成GO,然后加入解螺旋单链DNA,最后加入水合肼还原得到DNA功能化石墨烯。这其中主要是利用单链DNA与石墨烯之间的氢键以及静电作用对石墨烯进行功能化改性。
3 石墨烯基功能化复合材料
自2006年Stankovich等[34]首次报道了聚苯乙烯-石墨烯复合材料的制备方法后,石墨烯复合材料就迅速成为诸多领域的研究热点。以氧化石墨为前驱体,通过各种方法剥离还原得到具有良好分散性的石墨烯;再通过复合第二种成分,制备得到石墨烯复合材料[35, 36]。该类复合材料兼顾了两者的优异性能,在储能领域有较好的应用前景。Li等[37]通过Diels-Alder反应将石墨烯与四嗪衍生物反应后合成新聚吡咯-石墨烯片纳米复合材料,然后将该复合材料涂覆在电极表面,发现含40(wt)%聚吡咯的石墨烯纳米复合材料表现出最佳的电化学性能,0.5及2 A/g电流密度下的比电容分别为326和250 F/g。
Birrozzi等[38]以二氯化二苄基锡为原料,通过微波法还原分散在乙二醇中的聚丙烯酸(PAA)官能化GO,制得石墨烯/SnO2纳米复合阳极。在500mA/g电流密度下,循环140次后的电极具有430mAh/g的可逆比容量,库伦效率接近100%。这与PAA有效地确保SnO2在石墨烯基底中的良好分散密切相关。
Bandyopadhyay等[39]在GO的存在下,引发苯胺和间氨基苯磺酸的聚合制备磺化聚苯胺(SPANI),然后用水合肼还原得到功能化rGO复合材料(SPANI/G-1)。SPANI中磺酸基团的存在加速了氧化还原反应的进行,抑制了石墨烯的重新堆叠,并提供了赝电容。SPANI/G-1在1A/g的电流密度下比电容为1107F/g。SPANI/G-1中氧含量的减少使得石墨烯上发生sp2杂化的碳的数量增加,增强了石墨烯与SPANI链之间的π-π相互作用,促进了电子转移并且改善SPANI/G-1的电化学性质。使用SPANI/G-1作为正极和rGO作为负极的不对称超级电容器(ASC)在1.5A/g的电流密度下的比电容为157F/g。所设计的器件具有良好的倍率性能,在3.8A/g的电流密度下循环5000次后,仍有94%的容量保持率。此外,其能量密度和功率密度分别高达31.4Wh/kg和14764W/kg。
Sandra等[40]采用低温电化学法将石墨烯泡沫电沉积在Ni(OH)2上形成功能化石墨烯泡沫。该方法结合了单电极石墨烯优越的机械性能、电性能以及泡沫体的大比表面积和氢氧化镍的大赝电容等优点。电沉积后不需要进一步的热退火便可得到较大的比电容(900F/g),比无官能化石墨烯泡沫高两个数量级。
Kim等[41]将石墨烯和介孔SiO2复合体作为锂硫电池的骨架材料。通过将石墨烯、有序多孔的SiO2和嵌段共聚物前体的三元协同复合制备出功能化石墨烯片,如图 3所示。研究表明,复合物独特的介孔结构使其兼具石墨烯高电子电导率和SiO2作为聚硫原位吸收剂的双重功能,使锂硫电池具有良好的容量保持率和倍率性能。
Maroni等[42]在环保、低成本溶剂中,将Si纳米粒子分散在GO溶液中,预先采用低分子量PAA进行官能化,通过对悬浮液微波辐射诱导还原GO,采用过滤热退火的方法得到Si/rGO纳米复合材料(如图 4)。当采用高分子量的PAA作为粘合剂、碳酸亚乙烯酯作为电解液添加剂时,制备的复合正极材料表现出高的和稳定的可逆容量,可逆比容量为1000mAh/g。电流密度为500mA/g的条件下循环100次后,正极放电容量保持率约为80%。
Yu等[43]将负载Pd纳米颗粒的功能化石墨烯气凝胶(GA)用来作为一种高效的非对称超级电容器(ASCs)阳极材料。高表面积(328m2/g)和低的电阻率(比不含钯低50倍)赋予GA复合材料高的比电容(175.8F/g,5mV/s)、优良的倍率性能(扫描速率的10倍后,48.3%的保留)以及显著的可逆性。MnO2(阴极)和GA复合材料(阳极)组装的ASCs表现出快速充放电性能、优异的循环稳定性(3000次循环后89.6%保留)以及高的能量和功率密度(平均为13.9Wh/kg和13.3kW/kg)。
Xie等[36]通过熔融扩散法合成功能化凹凸棒石(ATTP)/石墨烯纳米片/硫复合材料(ATTP@GNs/S),并将其作为Li-S电池的正极。由于天然ATTP纳米纤维的强吸附能力和部分还原的石墨烯纳米片限制了锂多硫化物的溶出,ATTP/S和ATTP@GNs/S与单质硫相比电化学性能有所改善。在0.1C倍率下,复合材料的首次放电比容量为1143.9mAh/g,循环100次后可逆容量为512.0mAh/g,每个循环的容量衰减率低于0.5%。
Majumdar等[44]以石墨烯粉末为原料,采用了一种简单、绿色的超声辅助湿化学方法合成了羟基功能化石墨烯/MnO2纳米复合材料,用作超级电容器电极材料时,在10A/g的电流密度下循环1000次后,电容保持率约为91%,具有优异的比电容和循环稳定性。
4 结语
综上所述,功能化石墨烯可以调控其分子结构、电子能级和化学性质,从而实现石墨烯基材料的多元化应用。通过共价键功能化可以提高石墨烯的液相可加工性,制备的石墨烯材料比较稳定,并能够在保持石墨烯本征属性的同时赋予其新的性能;非共价键功能化操作简单、条件温和、对石墨烯的结构破坏非常小,可以最大程度地保留石墨烯的本征特性;石墨烯基功能化复合材料可以兼顾石墨烯和复合成分两者的优异性能,在储能领域有较高的应用前景。
但是,对石墨烯进行功能化会引入其他组分,改变石墨烯的本征结构,使得石墨烯的物理化学性质发生改变。因此,在对石墨烯进行功能化改性时,要尽可能的保持石墨烯的本征结构,控制功能化位点以及功能化基团的数量,并不断完善现有的功能化方法以及开发一些新的方法,充分发挥石墨烯的优异性能。另一方面,合成石墨烯基功能化复合材料的制造步骤以及该过程是否可以扩展也是需要考虑的。此外,石墨烯表面修饰的聚合物结构的多样性,将拓展其在储能以及其他领域的应用。
-
-
[1]
张谦, 张玲, 李景虹. 分析化学, 2013, 41: 641~649. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=fxhx201305003&dbname=CJFD&dbcode=CJFQ
-
[2]
杨蓉, 王黎晴, 吕梦妮等. 化学通报, 2016, 79: 503~508. http://www.hxtb.org/ch/reader/view_abstract.aspx?flag=1&file_no=20151229002&journal_id=hxtb
-
[3]
A A Komlev, T L Makarova, E Lahderanta et al. J. Magn. Magn. Mater. 2016, 415:45~50.
-
[4]
Y Li, Z Jian, M Lang et al. ACS Appl. Mater. Interf., 2016, 8:17352~17359. http://www.ncbi.nlm.nih.gov/pubmed/27328986
-
[5]
Z Sun, C L Pint, D C Marcano et al. Nat. Commun., 2011, 2:559. http://www.ncbi.nlm.nih.gov/pubmed/22127055
-
[6]
Y Zhao, X G Li, X Zhou et al. Sens. Actuat. B, 2016, 231:324~340.
-
[7]
Y Jing, X Yuan, Q Yuan et al. Sci. Rep., 2016, 6:29230.
-
[8]
V Georgakilas, M Otyepka, A B Bourlinos et al. Chem. Rev., 2012, 112:6156~6214.
-
[9]
A Wang, W Yu, Z Huang et al. Sci. Rep., 2016, 6:23325.
-
[10]
X Wang, F Zhang, J Xia et al. J. Electroanal. Chem., 2015, 738:203~208. http://www.sciencedirect.com/science/article/pii/S1572665714005347
-
[11]
J Ye, M T Ong, T W Heo et al. Sci. Rep., 2015, 5:16190. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4633639/
-
[12]
吕鹏, 冯奕钰, 张学全. 中国科学, 2010, 1247~1256. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=jexk201011002&dbname=CJFD&dbcode=CJFQ
-
[13]
艾伟. 南京邮电大学硕士学位论文, 2013.
-
[14]
孙宾宾. 广州化工, 2016, 23: 11~12. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=gzha201601006&dbname=CJFD&dbcode=CJFQ
-
[15]
E Jokar, S Shahrokhian, A I Zad. Electrochim. Acta, 2014, 147:136~142. http://www.sciencedirect.com/science/article/pii/S0013468614019331
-
[16]
杜祝祝. 南京邮电大学硕士学位论文, 2014. http://cdmd.cnki.com.cn/Article/CDMD-10293-1015554130.htm
-
[17]
刘跃文, 邓顺柳, 谢素原. 厦门大学学报, 2014, 53: 674~681. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=xdzk201405009&dbname=CJFD&dbcode=CJFQ
-
[18]
汪丽丽, 邢瑞光, 张邦文. 物理化学学报, 2014, 30(9): 1659~1666. doi: 10.3866/pku.whxb201406162
-
[19]
刘赞辉. 湖南大学硕士学位论文, 2013.
-
[20]
B Song, J Zhao, M Wang et al. Nano Energy, 2017, 31:183~193.
-
[21]
B Song, I C Ji, Y Zhu et al. Chem. Mater., 2016, 28(24):9110~9121.
-
[22]
Y Liu, Y Ma, S Guang et al. Carbon, 2015, 83:79~89. doi: 10.1016/j.carbon.2014.11.026
-
[23]
I Cha, E J Lee, H S Park et al. Synth. Met., 2014, 195:162~166. doi: 10.1016/j.synthmet.2014.05.019
-
[24]
H Qiu, X Han, F Qiu et al. Appl. Surf. Sci., 2016, 376:261~268. doi: 10.1016/j.apsusc.2016.03.018
-
[25]
N Lingappan, D W Kim, X T Cao et al. J. Alloys Compd., 2015, 640:267~274. doi: 10.1016/j.jallcom.2015.04.045
-
[26]
Z Xiang, Q Dai, J F Chen et al. Adv. Mater., 2016, 28:6253~6261.
-
[27]
王欢文. 西北师范大学硕士学位论文, 2011.
-
[28]
Y Han, M Shen, Y Wu et al. Synth. Met., 2013, 172:21~27. doi: 10.1016/j.synthmet.2013.04.001
-
[29]
S Fan, J Yang, T Wei et al. Talanta, 2016, 160:713~720. doi: 10.1016/j.talanta.2016.08.018
-
[30]
J J Shao, S D Wu, S B Zhang et al. Chem. Commun., 2011, 47:5771~5773. doi: 10.1039/c1cc11166c
-
[31]
D S Yu, T Kuila, N H Kim et al. Carbon, 2013, 54:310~322. doi: 10.1016/j.carbon.2012.11.043
-
[32]
X Yang, X Zhang, Z Liu et al. J. Phys. Chem. C, 2008, 112:17554~17558.
-
[33]
A J Patil, J L Vickery, T B Scott et al. Adv. Mater., 2009, 21:3159~3164. doi: 10.1002/adma.200803633
-
[34]
S Stankovich, D A Dikin, G H Dommett et al. Nature, 2006, 442:282~286.
-
[35]
H Jung, K T Park, M N Gueye et al. Int. J. Hydrogen Energy, 2016, 41:5019~5027. doi: 10.1016/j.ijhydene.2015.12.016
-
[36]
Q Xie, A Zheng, C Xie et al. Microp. Mesop. Mater., 2016, 224:239~244.
-
[37]
Y Li, G Louarn, P H Aubert et al. Carbon, 2016, 105:510~520. doi: 10.1016/j.carbon.2016.04.067
-
[38]
A Birrozzi, R Raccichini, F Nobili et al. Electrochim. Acta, 2014, 137:228~234. doi: 10.1016/j.electacta.2014.06.024
-
[39]
P Bandyopadhyay, T Kuila, J Balamurugan et al. Chem. Eng. J., 2017, 308:1174~1184. doi: 10.1016/j.cej.2016.10.015
-
[40]
S Ruiz Gómez, A Boscá, L Pérez et al. Diamond Relat. Mater., 2015, 57:63~67. doi: 10.1016/j.diamond.2015.03.003
-
[41]
K H Kim, Y S Jun, J A Gerbec et al. Carbon, 2014, 69:543~551. doi: 10.1016/j.carbon.2013.12.065
-
[42]
F Maroni, R Raccichini, A Birrozzi et al. J. Power Sources, 2014, 269:873~882. doi: 10.1016/j.jpowsour.2014.07.064
-
[43]
Z Yu, M McInnis, J Calderon et al. Nano Energy, 2015, 11:611~620. doi: 10.1016/j.nanoen.2014.11.030
-
[44]
D Majumdar, S Kumar, B Hattacharya. Mater. Today:Proceed., 2016, 3:3872~3877.
-
[1]
-

-



 下载:
下载:



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
