 图式1
二咪唑环糊精催化反应机理[8]
图式1.
Catalyse reaction mechanism of di-imidazole cyclodextrin[8]
图式1
二咪唑环糊精催化反应机理[8]
图式1.
Catalyse reaction mechanism of di-imidazole cyclodextrin[8]
引用本文:
罗莎杰, 王妍媖, 饶含兵, 王显祥. 人工模拟酶的研究与应用进展[J]. 化学通报,
2017, 80(7): 642-650.

Citation: Luo Shajie, Wang Yanying, Rao Hanbing, Wang Xianxiang. Advances in Research and Application of Artificial Enzymes[J]. Chemistry, 2017, 80(7): 642-650.
Citation: Luo Shajie, Wang Yanying, Rao Hanbing, Wang Xianxiang. Advances in Research and Application of Artificial Enzymes[J]. Chemistry, 2017, 80(7): 642-650.
人工模拟酶的研究与应用进展
English
Advances in Research and Application of Artificial Enzymes
Abstract:
Owning to its good stability, easy preparation, high environmental tolerance, artificial enzymes had been used widely to improve the activity and the yield of natural enzymes. In this paper, according to the classification of artificial enzymes, the present progresses in the study of traditional mimic enzymes and nanomaterials mimic enzymes had been reviewed. The advantages and disadvantages of the artificial enzymes were discussed in detail, and its prospects were also described.
-
Key words:
- Artificial Enzymes
- / Classification
- / Advantages and Disadvantages
-
自然界一切生命现象,比如生物体的生长发育、遗传、繁殖、运动、神经传导等都与酶催化过程紧密相关。可以说,如果没有酶,一切生命活动都将停止。天然酶的生物成分是几十甚至上千个氨基酸构成的生物大分子或具有催化功能的RNA(核酶)[1]。酶催化具有高效、专一、反应条件温和等特性,广泛应用于化学工程、医疗医药、食品卫生和农业生产等各个领域。然而,天然酶分离纯化难、成本高、价格贵以及稳定性差,难以满足现代工业的需求。因而,迫切需要稳定性好及易于制备的模拟酶来满足现代工业的要求。目前,模拟酶的研究取得了很多成果,开发了不同类型的模拟酶,如环糊精模拟酶、卟啉类模拟酶、纳米材料模拟酶等,已经在生物、医药、化工等领域得到了广泛的应用。本文将模拟酶分为传统模拟酶、纳米材料模拟酶两大类,对它们的研究进展和优缺点分别进行比较介绍。
1 传统模拟酶
模拟酶的化学成分是非蛋白类,但具有与天然酶相似的催化性能,其理论基础(主-客体化学[2]和超分子化学[3]理论)主要是由诺贝尔奖获得者Cram、Pederson与Lehn共同提出的。主-客体化学的基本原理来源于酶和底物之间的相互作用,即主体和客体在结合部位的空间及电子排列的互补。这种互补作用与酶和它所识别的底物结合情况类似。超分子化学理论的形成源于底物和受体的结合,这种结合主要靠氢键、范德华力及静电力等非共价键来维持。接受体与络合离子或分子结合,形成具有稳定结构和性质的实体,即形成“超分子”,它具有高效催化、分子识别及选择性输出等功能。根据酶催化反应机理,若合成出既能识别底物分子又具有酶活性部位催化基团的主体分子,同时主体分子能与底物发生多种分子间相互作用,那就能有效地模拟酶分子的催化过程[4]。传统模拟酶主要有三种模拟方式:(1) 模拟酶含有与天然酶相同的金属离子;(2) 模拟天然酶的活性中心结构;(3) 天然酶的整体模拟[5]。
根据主-客体化学和超分子理论,研究者们研究出了多种传统模拟酶。传统模拟酶不仅在耐酸碱、热稳定性方面优于天然酶,而且价格便宜,可大量应用于实际生产中。以下对几种重要的传统模拟酶进行分类介绍。
1.1 环糊精模拟酶
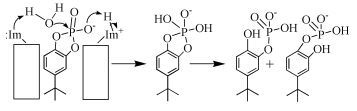
环糊精是由数个D-呋喃葡萄糖残基通过α-1, 4糖苷键连接而成的环状糖,每个残基均取无扭变变形的椅式构象。环糊精具有内部疏水、外部亲水的特性,能够更好地包络客体分子,对客体分子具有一定的选择性,是研究模拟酶很好的材料。1972年,Breslow等[6]用天然β-环糊精对人工酶进行研究,并设计了核糖核酸酶的模型,引起了大量研究者的关注。Bender等[7]发现,环糊精可以催化乙酸硝基酚酯的水解。单体环糊精虽具有催化性,但是其活性不高,之后研究者们对天然环糊精进行了进一步的研究,采用合成官能团修饰环糊精、与金属形成配合物、环糊精聚合物等方法提高模拟酶的催化活性。Breslow等[8]将两个咪唑基团同时引入环糊精的同一面,设计得到核糖核酸酶模型。该模型在催化4-叔丁基苯邻二酚环磷酯水解的过程中,两个基团具有协同作用,能更好地发挥催化作用,其催化过程示意图如图式1所示。
沈静如等[9]采用β-环糊精二间羧基苯磺酸酯同三氯化铁构建环糊精配合物模拟酶来同时模拟葡萄糖氧化酶和过氧化物酶,对葡萄糖制剂中的葡萄糖进行测定,线性范围为120~400 mg/L,检测下限为60mg/L,相对标准偏差(RSD)为1.9%(n=8)。Al-Maksoud等[10]合成了甲基化-β-环糊精,在过渡金属Pd存在下,其可催化丙烯醛缩二乙醇的芳基化反应。Toda等[11]将二氢尼克酰胺共价键合到β-环糊精的伯羟基处,用于模拟脱氧酶和氢转移酶。由于空腔内能够结合茚三酮,提高了还原基附近的底物浓度,在pH为7.0时,还原茚三酮的反应速率比二氢烟酰胺单独存在时快400倍。刘俊秋等[12]合成了2-位硒桥联环糊精模拟谷胱甘肽过氧化物酶,实验结果表明,该模拟物的酶活性比其他小分子模拟酶明显提高。Zhou等[13]利用锌离子环糊精配合物模拟酯水解酶,分别催化了双(4-硝基苯酚)碳酸酯、4-硝基苯酚乙酸酯、双(4-硝基苯酚)磷酸酯(BNPP)的水解反应,其中水解BNPP的Kcat值达到9.9×10-4 m-1·s-1,催化效果理想。Letort等[14]向甲基化β-环糊精中引入咪唑类和亚碘酰苯甲酸类基团模拟水解酶,其能够催化水解有机磷类毒剂。在催化反应中,引入的基团对有机磷类毒剂进行亲核攻击,提高了水解效率。此报道为环糊精在环境治理方面开辟了新的应用方向。
1.2 卟啉类模拟酶
卟啉是一类由四个吡咯类亚基的α-碳原子通过次甲基桥互联而形成的大分子杂环化合物,其主体为卟吩,有取代基的卟吩叫卟啉。卟啉上的两个吡咯质子被金属离子取代后,即为金属卟啉[15],根据取代基和取代金属离子的不同可以得到不同功能的配合物。金属卟啉及以金属卟啉为基础的衍生物一直是模拟酶研究的热点,它们以共轭大π键电子为体系,具有金属价态可变为基础稳定态的氧化还原特性;与此同时,其中心金属具有较强的配位能力[16]。
Groves等[17]报道用铁和锰的四苯基卟啉配合物模拟单加氧酶细胞色素P450,在常温常压下催化PhIO、NaClO、H2O2等氧化剂将烷烃和烯烃分别氧化成醇和环氧化物。1990年,Eills等[18]利用氯代苯基金属卟啉在室温下催化氧气直接氧化异丁烷为异丁醇,转化数>13000,选择性>90%。研究结果表明,卤代苯基金属为卟啉的外环取代基时的催化活性比没有取代苯基的金属卟啉的高。1995年,Murahashi等[19]研究发现,五氟苯基锰卟啉、钌卟啉以及钴卟啉能催化氧气氧化环己烷得到环己醇,在常压、温度为70℃时其催化活性很高,转化数最高可达17000。田恒水等[20]研究了四苯基卟啉、四-4-甲氧基苯基卟啉、四-4-氟苯基卟啉及3种卟啉相对应的钴、锰、铁金属卟啉在不同条件下催化氧气氧化丙烯生成环氧丙烷的反应。以四-4-氟苯基铁卟啉为催化剂时,在优化条件下,环氧丙烷的收率为40.38%,丙烯转化率为47.09%,环氧丙烷选择性高达85.75%。
卟啉配合物还可用于检测许多重要的生物分子。Motsenbocker等[21]利用牛血清白蛋白(BSA)和卟啉之间的相互作用,在抗体上进行锰卟啉修饰,得到了抗体-BSA-卟啉复合物,可应用于化学发光免疫分析中。姚凤姬等[22]研究了锰-四磺基苯卟啉络合物(Mn-TPPS4) 模拟辣根过氧化物酶(HRP)催化过氧化氢与邻苯二胺反应的特性,并与葡萄糖氧化酶的酶促反应偶联,用于测定血清中的葡萄糖。于雪花[23]通过导电聚合物聚3, 4-乙撑二氧噻吩(PEDOT)修饰玻碳电极,结合1-芘丁酸(PBA)制备了一种新的基于羟基铁卟啉类化合物(Hematin)的仿生酶传感器。该仿生传感器的制备过程简单、性质稳定,且对邻苯二酚和过氧化氢都有较高的检测灵敏度。
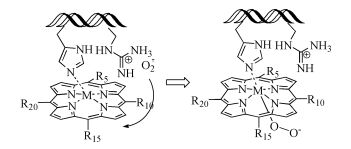
李刚等2[24]将两类水难溶性金属卟啉meso-四苯基卟啉金属配合物(MTPP,M=ZnⅡ,CoⅡ)和meso-四(p-羟基苯基)卟啉金属配合物(MTpHPP,M=ZnⅡ,CoⅡ)分别与BSA结合,制得水溶性金属卟啉白蛋白结合体(MP@BSA)。NBT光还原法测试表明MP@BSA具有清除超氧阴离子自由基(
${\rm{O}}_2^{\bar \cdot }$ )的性能。清除的机理是,BSA为小分子金属卟啉提供了良好的疏水环境,与此同时,白蛋白上带有正电荷的氨基酸残基具有吸引${\rm{O}}_2^{\bar \cdot }$ 的能力,有助于氧化-还原反应的电子传递,反应机理示意图见图式2。与小分子卟啉相比,MP@BSA的抗${\rm{O}}_2^{\bar \cdot }$ 性能得到了显著提升。对比单纯的VC或BSA,其氧化活性亦提高了很多倍。其中,羟基取代金属卟啉结合体表现出更为优良的清除${\rm{O}}_2^{\bar \cdot }$ 特性,CoTpHPP@BSA的半最大效应浓度为1.5μmol/L,对天然Cu,ZnSOD的模拟度为2.73%。Chapmana等[25]研究了铁卟啉催化的对乙酰氨基酚前药的生物模拟氧化,考察了含氮和硫醇的螯合配体对药物及代谢物形成的影响。雷建平等[26]发现,DNA-铁卟啉复合物能够有效地模拟过氧化物酶,铁卟啉可以镶嵌在DNA的沟槽内,形成DNA-铁卟啉复合物,改善了铁卟啉易于团聚的缺陷,有效提高了其催化活性。他们还应用DNA-铁卟啉复合物作为信号标签,建立了一种荧光放大检测DNA的方法。该方法检测DNA具有高的灵敏度和特异性。
1.3 分子印迹聚合物模拟酶
以印迹分子为模板与功能单体在交联剂的作用下相结合制备成分子印迹聚合物(MIP),然后将印迹分子从MIP中除去,留下能与印迹分子相特异结合的分子结构[27]。因此,得到的MIP具有对印迹分子及其相似结构的一类分子特异性识别的功能。1972年,Wulff等[28~29]首次成功制备了共价型MIP,此MIP对糖类化合物有较高的选择性;随后,Mosbach等[30]开发了非共价型MIP制备方法。此后,分子印迹技术得到迅速发展。MIP作为模拟酶可以用于催化不对称催化合成、环加成反应、酯的水解与醇解、碳-碳键的形成[31]等。Toorisaka等[32]用烷基咪唑和钴离子形成的络合物作为模板分子合成的MIP可用于催化水解氨基酸酯,其对底物表现出特异亲和性。Sellergren等[33]设计了一系列模板分子,目的是将糜蛋白酶的关键催化元素引入聚合物活性位点,模拟蛋白水解酶的活性中心。当模板分子为D构型时,MIP能够选择性催化D构型N-叔丁氧基羰基苯丙氨酸对硝基苯酯的水解反应。王红飞等[34]以马来酸酐酯化葡聚糖与氨基吡啶偶联物(Dex-MA-AP)为功能单体、过渡态类似物p-硝基苯磷酸酯(NPP)为模板分子、Co2+为配位中心离子,通过反相乳液法制得对p-硝基乙酸苯酯(NPA)水解反应具有催化活性的分子印迹微凝胶模拟酶。结果显示,此模拟酶不仅对NPA的水解反应有非常高的催化特性,而且对底物表现出明显的催化选择,其催化NPA水解最大反应速率为2.51×10-8 mol·h-1。
1.4 胶束类模拟酶
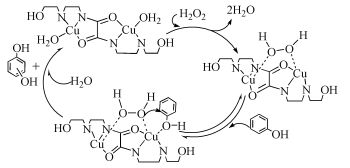
表面活性剂在水溶液中超过一定浓度后能自发聚集成胶束。胶束可以为底物分子提供一个疏水环境,对底物分子具有包络作用,与酶的结合部位类似。若再将某些作用基团如咪唑、硫醇、羟基及一些辅酶连接到胶束上面,就能够制备具有催化活性的模拟酶。Tonellato等[35]将Cu2+与胶束以1:1的比例形成配合物模拟羧肽酶A催化α-吡啶甲酸对硝基苯酚酯(PNPP)的水解。谢家庆等[36]将铜的双核配合物与表面活性剂聚氧乙烯月桂醚或月桂酰肌氨酸钠合成金属胶束模拟过氧化物酶,其可催化H2O2氧化苯酚的反应。由于两个铜离子的协同作用,金属胶束(ML)与H2O2能够快速生成活化络合物,再与苯酚生成反应中间体。中间体的形成,使电子由分子间的传递转化为分子内传递,由此降低的反应的活化能,使反应速率增加,催化机理如图式3所示。该反应受ML的用量、反应温度和pH的影响。
1.5 其他传统模拟酶
除以上介绍的几种重要的传统模拟酶外,冠醚类、杯芳烃类、单核及双核配合物模拟酶亦受到人们的关注。
冠醚是一种简单的环状化合物,可以通过不同的合成方法调节其空腔大小,从而选择性地与金属离子、有机或无机小分子络合形成稳定的化合物[37]。胡伟等[38]用杂氮冠醚化席夫碱钴(Ⅱ)配合物,模拟生物体内的磷酸二酯水解酶的催化作用。两种氮杂冠醚单席夫碱钴(Ⅱ)配合物均对BNPP水解表现出很好的催化活性。
杯芳烃是由苯酚基和亚甲基或类似基团交替连接形成的环状低聚物。与冠醚相似,杯芳烃也具有可调节的疏水空腔,可以对金属离子和中性分子进行包络[39]。袁立华等[40]将带有疏水空腔的对叔丁基杯[4]芳烃与卟啉羧酸的酰氯化物反应,并导入产生酶活性的辅因子,合成了仿细胞色素P-450单加氧酶模型物,经表征确定模型物中杯环呈现锥体构象。Molenveld等[41]合成了含铜的双核杯[4]化合物,成功地模拟了DNA水解酶对磷酸二酯键的作用。
单核及双核配合物模拟酶主要对金属酶进行模拟。大多数金属酶的活性中心都含有金属离子,这与配位化合物相似。因此,通过对配体的修饰可以合成出与天然酶相似的配合物[16]。朱兵等[42]将稀土双核配合物用于催化剪切环状质粒pCAT及pCUC19,使之变为线性DNA链,成功模拟生物体内的核酸酶。梅光泉等[43]合成了三种锰(Ⅱ)配合物,对其清除活性氧的催化性能进行研究,证明该配合物具有超氧化物歧化酶的性能。
相比天然酶,传统模拟酶在耐酸、耐碱、热稳定性等方面都具有优势,而且价格便宜,能大规模用于实际应用中。但是,传统模拟酶也存在合成较为复杂、催化活性位点单一、催化效率低以及分离、回收和再生较困难等缺点。
2 纳米材料模拟酶
纳米粒子是一类大小在1~100 nm之间、具有独特的化学、电学、物理、力学和机械性能的粒子。由于纳米粒子具有尺寸效应、表面与界面效应和宏观量子隧道效应等不同于常规材料的特性,使其在农业生产、汽车工业、食品加工、药物传递、电子产品、医学成像、化妆品、分析检测、建筑材料等领域得到了广泛应用[44~45]。2007年,Gao等[46]发现Fe3O4纳米粒子具有HRP模拟酶作用,打破了以往认为纳米材料的生物惰性的认识。近年来,随着对纳米材料研究的深入,大量文献报道证实纳米材料能够模拟多种天然酶活性。
2.1 Fe3O4磁性纳米材料
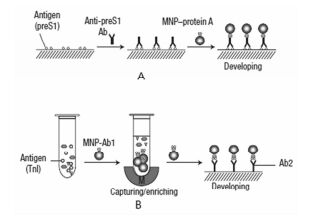
由于Fe3O4纳米粒子具有独特的磁性和生物相容性,备受各领域研究者们的青睐。大多数研究集中在其磁性的应用,如磁性分离、医学诊断、药物传递等。2007年,Gao等[46]发现,Fe3O4磁性纳米颗粒(MNPs)能够模拟HRP,在过氧化氢存在的情况下,能够氧化HRP底物,如氧化3, 3′, 5, 5′-四甲基联苯胺(TMB)呈现蓝色,氧化2, 2′-联氮双二铵盐(ABTS)呈现绿色。其催化特性与HRP一致,符合酶催化的动力学。Fe3O4MNPs对底物的亲和力高于天然酶HRP,能承受高浓度过氧化氢,且能对抗恶劣的环境而不易失活。他们将Fe3O4MNPs用于免疫检测,首先在模板上包覆乙肝病毒(PreS1),然后将它与乙肝病毒抗体一起孵育,再将固定有蛋白A的Fe3O4MNPs与TMB一起加入,再通过检测Fe3O4催化TMB显色的强度检测乙肝表病毒,如图 1(a)所示。类似地,还可以用于检测心肌肌钙蛋白(TnI),如图 1(b)所示。
Wei等[47]用Fe3O4MNPs模拟HRP,用于检测葡萄糖。由于葡萄糖在葡萄糖氧化酶的催化下能产生H2O2,故可以通过检测H2O2来间接检测葡萄糖,其对H2O2的检测限达到3×10-6mol/L,线性检测范围为5×10-6~1×10-4 mol/L,且对葡萄糖的检测具有专一性。Wang等[48]将Fe3O4MNPs用于检测四环素类抗生素。由于四环素类抗生素可以与Fe3O4MNPs形成络合物,能够提高Fe3O4MNPs本身的过氧化物酶催化速率,从而可实现检测四环素。Fe3O4MNPs对不同四环素具有不同的检测限,其中对土霉素的检测限达到26nmol/L。该方法与常规方法相比,操作简单、成本低、选择性高,可以用于临床药物分析。Chen等[49]利用新的方法合成Fe3O4MNPs,其可催化H2O2降解废水中有机污染物若丹明B,对若丹明B的降解率在2h内达到98%。此外,该Fe3O4MNPs对其他有机污染物如亚甲基蓝、四环素、环丙沙星、橙黄G等也具有催化降解作用。类似地,Fe3O4MNPs还可以用于检测胆固醇[50]、肾上腺素[51]、胆碱[52]、还原性谷胱甘肽[53]、核酸[54]等;此外,还可以用于病原微生物抑制[55]、磺胺降解[56]、若丹明降解[57]、肿瘤细胞抑制[58]、苯酚和苯胺化合物的降解[59]等。
实际上,Fe3O4MNPs的粒径[60]、形状[61]及不同的表面修饰[62]均会影响粒子的催化活性,这可能是由于粒子催化活性与粒子表面暴露的铁原子多少及表面电荷分布密切相关。此外,Fe3O4MNPs本身具有超顺磁性,能够实现快速分离与回收再利用;并且,将其与具有其他性质的物质结合,可得到多功能复合材料。
Fe3O4-金-氧化硅复合材料可同时模拟HRP和葡萄糖氧化酶,通过比色检测葡萄糖浓度,最低检测限为5×10-7mol/L,线性范围为1×10-5~1.3×10-4mol/L[63]。Fe3O4-还原氧化石墨烯复合材料(Fe3O4NSs/rGO)改善了Fe3O4本身的容易聚集、不稳定等缺陷,使复合材料具有更大的比表面积和更高的稳定性[64]。酶动力学研究表明,Fe3O4NSs/rGO对H2O2的Km为0.25mmol/L,相比Fe3O4MNPs(Km=154mmol/L)[46]具有更高的亲和力及更广泛的应用。Fe3O4NSs/rGO可以用于检测乙酰胆碱(ACh),ACh在乙酰胆碱酯酶(AChE)的作用下生成胆碱(choline),氧化酶(chox)进一步将胆碱氧化生成H2O2。Fe3O4NSs/rGO通过比色法检测生成的H2O2间接检测ACh。对ACh的检测限达到3.9×10-5mol/L,线性检测范围1×10-4~1×10-2mol/L。通过对牛奶样品中ACh的检测结果表明,该复合材料具有应用于食品检验的潜力。Zhang等[65]将银-金-Fe3O4复合物(Ag@Au-Fe3O4MNPs)建立的免疫传感器用于检测蛋白生物标志物。该免疫传感器对H2O2的还原反应表现出优良电化学催化活性,可用于检测免疫球蛋白(IgG),检测限为50fg/mL,检测线性范围0.1pg/mL~5μg/mL。此外,研究者们还合成了Fe3O4-铂复合材料用于免疫检测[66];Fe3O4-碳复合材料用于肿瘤细胞抑制[67];Fe3O4-Mn3O4/rGO复合物用于降解磺胺甲嘧啶[68];钯-Fe3O4复合物用于降解异丁苯丙酸(布洛芬)[69];Fe3O4-CeO2复合物用于降解儿茶酚[70]。
2.2 贵金属纳米材料
金属纳米粒子具有金属催化活性位点,在催化方面也有广泛应用。Jv等[71]和Jin等[72]利用带正电荷的Au团簇和BSA稳定的Au团簇(BSA-Au clusters)模拟过氧化物酶分别检测了过氧化氢和葡萄糖。对葡萄糖的最低检测限分别为4×10-6和5.0×10-6mol/L,线性检测范围分别为1.8×10-5~1.1×10-3和1.0×10-5~0.5×10-3 mol/L。Wang等[73]将BSA-Au clusters用于胰岛素的检测。BSA-Au clusters具有光催化作用,由于胰岛素可以水解BSA蛋白,降低BSA-Au clusters的光催化活性,从而通过测定其活性的强弱来检测胰岛素的含量。对胰岛素的检测限达到0.6μg/mL,检测范围为0.9μg/mL~1.0mg/mL。
此外,研究者们还将贵金属纳米材料与其他材料结合得到双功能复合材料,使其得到更好的应用。Xiong等[74]利用Au-Pt复合材料模拟超氧化物歧化酶和过氧化物酶,清除细胞内的自由基。该复合物表现很好的生物相容性,能够被人类皮肤上的细胞有效吸收,有望用于治疗自由基介导的疾病。Zhan等[75]用金-GO复合材料模拟过氧化物酶用于检测呼吸道合胞病毒(RSV)。检测的原理类似于酶联免疫吸附测定(ELISA),首先将RSV与RSV抗体一起孵育在反应板上,再加入AuNPs-GO与第二抗体结合得到的Ab-AuNPs-GO一起孵育,最后加入H2O2和TMB,通过比色法对RSV进行定量分析。Liu等[76]报道了一种碳基Au-Ag双金属纳米粒子,用于人免疫球蛋白(HIgG)的检测。碳基Au-Ag双金属纳米粒子在其中同时作为催化剂和信号增强剂,对HIgG具有很低的检测限(0.0009ng/mL),检测线性范围为0.005~100 ng/mL。此外,贵金属复合材料还可用于葡萄糖[77~78]、胆固醇[79]、汞离子、甲基汞[80]、核酸[81]、凝血酶[82~83]、肿瘤细胞[84]、猪繁殖与呼吸综合征病毒[85]、ACh[86]、瘦肉精[87]等的检测,有机污染物的降解[88]、病原微生物的抑制[89]等。
笔者课题组研究了超顺磁性Fe3O4纳米材料、荧光量子点和金团簇的制备及其在生物体中的应用。制备了Fe3O4@Au[90]用于固定化酶,Fe3O4@SiO2[91]用于DNA提纯;合成了CdX(X=Se,Te,Te/Zns)系列量子点,将其作为载体用于有毒金属离子的可视化检测[92],并探究了其作为一种荧光探针用于药物体内示踪的可能性[93]。在研究金团簇作为探针检测小分子[94~95]的同时我们首次发现,BSA-Au团簇能够用作模拟过氧化物酶,代替天然过氧化氢酶用于酶催化反应[96]。BSA-Au团簇在较宽的pH和温度范围都具有很高的酶活性,而且和其他纳米材料作为模拟化过氧化物酶相比,其对底物TMB的亲和性更高。在H2O2存在下,BSA-Au团簇能催化氧化TMB,产生有色物质,从而建立了检测H2O2的新方法,对H2O2检测的线性范围为5.0×10-7~2.0×10-5 mol/L,检出限为2.0×10-8 mol/L。黄嘌呤氧化酶能够专一性地催化黄嘌呤产生H2O2,我们根据该原理建立了间接检测生物样品中黄嘌呤的新方法,检测线性范围为1×10-6~2×10-4 mol/L,检出限为5×10-7mol/L。目前,笔者课题组正在重点从事多功能模拟酶材料Fe3O4@Au模拟酶、SiO2@BSA-Au团簇模拟酶、Fe3O4@SiO2@Au模拟酶等的制备及在生物小分子检测和环境催化降解中的应用研究。
2.3 碳基纳米材料
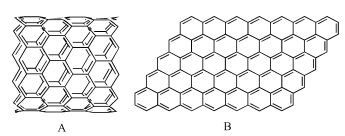
碳基纳米材料是指其基本单元至少有一维是小于100nm的碳材料。碳基纳米材料的种类比较多,石墨烯、碳纳米管、碳点都是现阶段研究的热点。石墨烯是Novoselov等[97]在2004年报道的一种新型材料,它由单原子层晶体构成,所有碳原子排列于蜂窝状结构单元中,其厚度为单个碳原子厚度,如图式4(B)。
 图式4
碳基纳米材料(A:碳纳米管;B:石墨烯)
图式4.
Carbon-based nano-materials (A: carbon nanotube; B:graphene)
图式4
碳基纳米材料(A:碳纳米管;B:石墨烯)
图式4.
Carbon-based nano-materials (A: carbon nanotube; B:graphene)
Song等[98]报道GO具有HRP活性,在H2O2的存在下能氧化TMB显蓝色,因此可以用于检测H2O2和葡萄糖。检测葡萄糖的线性范围为1×10-6~2×10-5 mol/L,最低检测限为1×10-6mol/L。Li等[99]报道一种rGO-铜复合物可超灵敏检测多巴胺,结合此复合物电催化与模拟酶催化的特性,建立了新型电化学仿生传感器,对多巴胺的检测限为3.48nmol/L,检测范围为0.01~40 μmol/L。此外,还有关于石墨烯用于检测谷胱甘肽[100]、石墨烯血红素复合物用于检测单核苷酸多态性[101]、石墨烯金复合物用于检测金属蛋白酶[102]的报道。
碳纳米管是由石墨层折叠而形成的无接缝的碳圆柱体,有多壁碳纳米管(MWNTs)和单壁碳纳米管(SWNTs),粒径一般为几十纳米至微米级,其长径比很大。Song等[103]发现SWNTs也具有HRP活性,通过实验证明催化性质来自碳纳米管本身,而非其他杂质。他们通过这种模拟酶的特性建立了一种检测与疾病相关的单核苷酸多态性(SNP)的方法,最低检测限达到1nmol/L。文中报道单链DNA(ssDNA)可以与碳纳米管形成稳定的复合物,而双链DNA(dsDNA)虽能与碳纳米管形成复合物,但相互作用力大大弱于ssDNA。在高盐的环境下,SWNTs的相互排斥力会减弱而发生聚集。ssDNA与SWNTs形成的复合体可以使SWNTs表面带有更强的负电荷而避免聚集,dsDNA却不能避免SWNTs的聚集。因此,通过离心,SWNTs与dsDNA形成的复合物会沉淀下来,再加入TMB和H2O2产生颜色反应,可实现对dsDNA进行分析。Cui等[104]运用水热-氢气还原法得到了具有过氧化氢酶活性的螺旋状碳纳米管,所建立的H2O2传感器检测限为5×10-4mol/L。
碳点是2004年报道的一种新型荧光纳米材料[105],它来源于蜡烛灰,不仅具有碳基纳米材料的特性,又具有传统量子点的发光特性。Qu等[106]发现碳点具有内在的HRP活性,基于此实现了对葡萄糖及真实生物样本的检测,最低检测限达到2×10-5mol/L。
2.4 其他纳米材料
除以上介绍的纳米材料外,还有其他一些纳米材料应用于天然酶的模拟。Asati等[107]报道CeO2纳米粒子具有氧化酶的活性,在中性条件下可用于检测肿瘤的生物标记。该方法操作简便且耗时短,为医学检验提供了一种新的可靠的检测方法。Mu等[108]研究发现,CeO2/NiO复合材料具有过氧化物酶的活性,铈含量为2.5%时表现出最高活性。其对H2O2的检测限为0.88μmol/L,检测范围为0.05~40 mmol/L。此外,CeO2纳米还具有过氧化物酶[109]、超氧化物歧化酶[110]的活性,分别用于检测葡萄糖、清除自由基。Mu等[111]研究发现Co3O4纳米粒子(Co3O4NPs)具有过氧化物酶的催化性能,将其用于检测H2O2和葡萄糖,检测葡萄糖的线性范围为1×10-5~1×10-2mol/L,检测限为5×10-6mol/L。Zhang等[112]报道普鲁士蓝-Co3O4复合材料(PB@Co3O4)具有过氧化物酶活性,将该复合物用于谷胱甘肽的检测,检测限为0.021μmol/L,检测范围0.1~10 μmol/L。类似地,Co3O4NPs还可应用于免疫检测[113]、钙检测[114]、亚硫酸盐的检测[115]等。
Dong等[116]报道铂纳米粒子具有尿酸酶的活性,能够催化尿酸的氧化降解,有望成为治疗痛风的有效药物。铂纳米粒子还具有过氧化物酶的活性,可用于葡萄糖[117]、汞离子[118]等的检测。
此外,研究者们还发现氧化铜[119]、硫化镉[120]、铁酸钴[121]、二氧化锰[122]等纳米材料均具有过氧化物酶活性。
3 结语
随着研究的不断深入,模拟酶已经得到了广泛的应用,特别是在生物小分子检测领域取得了巨大的成就。与天然酶相比,模拟酶具有性质稳定、易于制备、成本低、可重复使用、环境耐受性强等优点。目前模拟酶的研究主要集中在过氧化物酶、氧化酶、超氧化物歧化酶等少数几种酶的模拟,而自然界中的酶却有成千上万种,如何开发未知的模拟酶,是一个颇具挑战性的课题。除此之外,模拟酶催化效能偏低、催化性能有限、底物选择特异性不高等缺点仍需不断改进,酶的形态、大小、表面修饰等性质与模拟酶的催化性能的相互关系仍需进一步研究。因此酶的制备方法与后处理的优化是目前研究的重点之一。
综上所述,虽然模拟酶仍存在大量问题。但随着研究者们对模拟酶的性质、催化机制的深入研究及有效控制,以及与其他科学技术的结合,模拟酶技术有望在化学工程、生物医学、食品安全、环境卫生等领域得到广泛应用,为人类生活和社会发展做出巨大贡献。
-
-
[1]
王镜岩, 朱圣庚, 徐长法. 生物化学(上册). 北京:高等教育出版社, 2002:319~320.
-
[2]
D J Cram. Science, 1974, 183:803~809.
-
[3]
J M Lehn. Angew. Chem. Int. Ed., 1988, 27:89~112.
-
[4]
邢锦娟, 刘琳. 辽宁工业大学学报(自然科学版), 2009, 29(2):125~128.
-
[5]
王夔. 化学通报,1985, (7):1~8.
-
[6]
R Breslow. Chem. Soc. Rev, 1972, 1(4):553~580.
-
[7]
M L Bender, M Komiyama. Cyclodextrin Chemistry. Springer Science & Business Media, 2012.
-
[8]
R Breslow, J B Doherty, G Guillot et al. J. Am. Chem. Soc., 1978, 100:3227~3229.
-
[9]
沈静茹, 雷灼霖, 孙小梅等. 分析化学, 2001, 29(7):828~831.
-
[10]
W Al-Maksoud, S Menuel, M Jahjah et al. Appl. Catal. A, 2014, 469(17):250~258.
-
[11]
陆冬梅, 罗志刚, 杨连生等. 粮油食品, 2005, 13(4):27~28.
-
[12]
刘俊秋, 高姝娟, 罗贵民等. 中国生物化学与分子生物学报, 1999, 15(1):102~104.
-
[13]
Y H Zhou, M Zhao, Z W Mao et al. Chem. Eur. J., 2008, 14:7193~7201.
-
[14]
S Letort, D Mathiron, T Grel et al. Chem. Commun., 2015, 51:2601~2604.
-
[15]
徐洁. 南京大学硕士学位论文, 2013.
-
[16]
刘有芹, 颜芸, 沈含熙等. 化学进展, 2005, 17(6):1067~1073.
-
[17]
D Mansuy. Pure. Appl. Chem., 1987, 59(6):759~770.
-
[18]
P E Ellis, J E Lyons. Coord. Chem. Rev., 1990, 105:181~193.
-
[19]
S I Murahashi, T Naota, N Komiya. Tetrahed. Lett., 1995, 36(44):8059~8062.
-
[20]
赵付强, 田恒水. 化工进展, 2016, 35(2):519~523.
-
[21]
M Motsenbocker, J Y Ichimori, K Kondo. Anal. Chem., 1993, 65:397~402.
-
[22]
姚凤姬, 王林涛, 慈云祥. 北京大学学报(自然科学版), 1999, 35(4):437~440.
-
[23]
于雪花. 南京理工大学硕士学位论文, 2015.
-
[24]
殷晓春, 李刚, 王荣民等. 中国科学:化学, 2013, 43(2):171~177.
-
[25]
C M Chapmana, J M Pruneaua, C A Laveracka et al. Appl. Catal. A, 2016, 510:204~215.
-
[26]
N Xu, J P Lei, Q B Wang et al. Talanta, 2016, 150:661~665.
-
[27]
韩霜. 东北石油大学硕士学位论文, 2014.
-
[28]
G Wulff, A Sarhan. Angew. Chem. Int. Ed., 1972, 11:341~344.
-
[29]
G Wulff, A Sahan, K Zabrocki. Tetrahed. Lett., 1973, 44(4):4329~4332.
-
[30]
G Vlatakis, L I Andersson, R Muller et al. Nature, 1993, 361:645~647.
-
[31]
娄忠良, 孟子晖, 王鹏孟等. 有机化学, 2009, 29(11):1744~1749.
-
[32]
E Toorisaka, K Uezu, M Goto. Biochem. Eng. J., 2003, 14(2):85~91.
-
[33]
B Sellergren, R N Karmalkar, K J Shea. J. Org. Chem., 2000, 65(13):4009~4027.
-
[34]
王红飞, 唐春燕, 杨浩等. 高等学校化学学报, 2010, 31(12):2488~2493.
-
[35]
P Scrimin, P Tecilla, U Tonellata. J. Org. Chem., 1994, 59(1):18~24.
-
[36]
J Q Xie, G X Chen, H Yan et al. J. Dispers. Sci. Technol., 2007, 28:505~510.
-
[37]
张来新, 朱海云. 化学与生物工程, 2013, 30(5):5~7.
-
[38]
胡伟, 刘富安, 谢家庆等. 化学研究与应用, 2004, 16(2):215~218.
-
[39]
汪培承. 同济大学硕士学位论文, 2008.
-
[40]
袁立华, 陈淑华, 赵华明. 化学学报, 1994, 52:1035~1040.
-
[41]
P Molenveld, F J Engbersen, H Kooijman et al. J. Am. Chem. Soc., 1998, 120(27):6726~6737.
-
[42]
朱兵, 赵大庆, 倪嘉缵. 化学进展, 1998, 10(4):396~440.
-
[43]
梅光泉, 田亚平, 方允中等. 无机化学学报, 2002, 18(4):357~361.
-
[44]
R J Aitken, M Q Chaudhry, A B A Boxall et al. Occup. Med., 2006, 56:300~306.
-
[45]
C N R Rao, A K Cheetham. J. Mater. Chem., 2001, 11:2887~2894.
-
[46]
L Z Gao, J Zhang, L Nie et al. Nanotechnology, 2007, 2(10):577~583.
-
[47]
H Wei, E Wang. Anal. Chem., 2008, 80:2250~2254.
-
[48]
Y L Wang, Y J Sun, H C Dai et al. Sensor Actuat. B, 2016, 236:621~626.
-
[49]
F Chen, S Xie, X Huang et al. J. Hazard. Mater., 2017, 322:152~162.
-
[50]
M I Kim, J Shim, T Li et al. Chem. Eur. J., 2011, 17:10700~10707.
-
[51]
C Liu, C Yu, W Tseng. Anal. Chim. Acta, 2012, 745:143~148.
-
[52]
Z Zhang, X Wang, X Yang. Analyst, 2011, 136:4960~4965.
-
[53]
Y Ma, Z Zhang, C Ren et al. Analyst, 2012, 137:485~489.
-
[54]
K S Park, M I Kim, D Y Cho et al. Small, 2011, 7:1521~1525.
-
[55]
L Gao, K M Giglio, J L Nelson et al. Nanoscale, 2014, 6:2588~2593.
-
[56]
H Niu, D Zhang, S Zhang et al. J. Hazard. Mater., 2011, 190:559~565.
-
[57]
N Wang, L Zhu, M Wang et al. Ultrason. Sonochem., 2010, 17:78~83.
-
[58]
D Zhang, Y Zhao, Y Gao et al. J. Mater. Chem. B, 2013, 1:5100~5107.
-
[59]
S Zhang, X Zhao, H Niu et al. J. Hazard. Mater., 2009, 167:560.~566
-
[60]
F F Peng, Y Zhang, N Gu. Chem. Lett., 2008, 19, 730~733.
-
[61]
S Nath, C Kaittanis, V Ramachandran et al. Chem. Mater., 2009, 21, 1761~1767.
-
[62]
F Q Yu, Y Z Huang, A J Cole et al. Biomaterials, 2009, 30:4716~4722.
-
[63]
X He, L F Tan, D Chen et al. Chem. Commun., 2013, 49:4643~4645.
-
[64]
J Qian, X W Yang, L Jiang et al. Sensor. Actuat. B, 2014, 201:160~166.
-
[65]
H F Zhang, L Ma, P L Li et al. Biosens. Bioelectron., 2016, 83:343~350.
-
[66]
Z Yang, Y Q Chai, R Yuan et al. Sensor. Actuat. B, 2014, 193:461~466.
-
[67]
Q An, C Y Sun, D Li et al. ACS Appl. Mater. Int., 2013, 5:13248~13257.
-
[68]
Z Wan, J Wang. J. Hazard. Mater., 2017, 324:653~664.
-
[69]
B Thokchom, P P Qiu, M C Cui. Ultrason. Sonochem., 2017, 34:262~272.
-
[70]
A Gogoi, M Navgire, K C Sarma et al. Chem. Eng. J., 2017, 311:153~162.
-
[71]
Y Jv, B X Li, R Cao. Chem. Commun., 2010, 46:8017~8019.
-
[72]
L H Jin, L Shang, S J Guo et al. Biosens. Bioelectron., 2011, 26:1965~1969.
-
[73]
Wang G L, Jin L Y, Dong Y M et al. Biosens. Bioelectron., 2015, 64:523~529.
-
[74]
B Xiong, R L Xu, R Zhou et al. Talanta, 2014, 120:262~267.
-
[75]
L Zhan, C Li, W Wu et al. Chem. Commun., 2014, 50:11526~11528.
-
[76]
Z Si, R Li, X Liu et al. Biosens. Bioelectron., 2017, 92:457~464..
-
[77]
Y Li, Q Ma, Z Liu et al. Anal. Chim. Acta, 2014, 840:68~74.
-
[78]
X Z Zhang, Y Zhou, W Zhang et al. Colloid. Surf. A, 2015, 490:291~299.
-
[79]
J B Liu, X N Hu, S A Hou et al. Sensor Actuat. B, 2012, 166~167:708~714.
-
[80]
C W Tseng, H Y Chang, J Y Chang et al. Nanoscale, 2012, 4:6823~6830.
-
[81]
Y Zhang, Y F Zhang, C L Xu et al. Biosen. Bioelectron., 2013, 43:205~210.
-
[82]
Y Wang, Y Zhang, T Yan et al. Biosens. Bioelectron., 2016, 80(65):640~646.
-
[83]
C W Lien, C C Huang, H T Chang et al. Chem. Commun., 2012, 48:7952~7954.
-
[84]
Y Tao, Y Lin, Z Huang et al. Adv. Mater., 2013, 25:2594~2599.
-
[85]
K Shao, C J Zhang, S Y Ye et al. Sensor Actuat. B, 2017, 240:586~594.
-
[86]
M S Mathew, A Baksi, T Pradeep et al. Biosens. Bioelectron., 2016, 81:68~74.
-
[87]
Y Yang, H Zhang, C Huang et al. Biosens. Bioelectron., 2016, 89:461~467.
-
[88]
Y H Chiu, Y J Hsu. Nano Energy, 2017, 31:286~295.
-
[89]
Y Tao, E Ju, J Ren et al. Adv. Mater., 2015, 27:1097~1104.
-
[90]
X X Wang, S Huang, Z Shan et al. Chin. Sci. Bull., 2009,54:1176~1181.
-
[91]
X X Wang, S Wang, W S Yang et al. Acta Chim. Sin., 2009,67:54~58.
-
[92]
X Wang, Y Lv, X Hou. Talanta, 2011, 84:382~386.
-
[93]
X X Wang, Z K Yang, Z San et al. Sci. China Chem., 2010, 53:1718~1722.
-
[94]
X Wang, P Wu, Y Lv et al. Microchem. J., 2011, 99:327~331.
-
[95]
X Wang, Y Wang, H Rao et al. J. Brazil. Chem. Soc., 2012,23:2011~2015.
-
[96]
X X Wang, Q Wu, Z Shan et al. Biosens. Bioelectron., 2011, 26:3614~3619.
-
[97]
K S Novoselov, A K Geim, S V Morozov et al. Science, 2004, 306(5696):666~669.
-
[98]
Y J Song, K G Qu, C Zhao et al. Adv. Mater., 2010, 22(19):2206~2210.
-
[99]
Y Li, Y Gu, B Zheng et al. Talanta, 2016, 162:80~89.
-
[100]
A Zheng, Z Cong, J Wang et al. Biosens. Bioelectron., 2013, 49:519~524.
-
[101]
Y Guo, L Deng, J Li et al. ACS Nano, 2011, 5:1282~1290.
-
[102]
P D Nguyen, V T Cong, C Baek et al. Biosens. Bioelectron., 2015, 89:666~672.
-
[103]
Y J Song, X H Wang, C Zhao et al. Chem. Eur. J., 2010, 16(12):3617~3621.
-
[104]
R J Cui, Z D Han, J J Zhu. Chem. Eur. J., 2011, 17(34):9377~9384.
-
[105]
X Y Xu, R Ray, Y L Gu et al. J. Am. Chem. Soc., 2004, 126(40):12736~12737.
-
[106]
X H Wang, K G Qu, B L Xu et al. Nano. Res., 2011, 4(9):908~920.
-
[107]
A Asati, C Kaittanis, S Santra et al. Anal. Chem., 2011, 83:2547~2553.
-
[108]
J S Mu, X Zhao, J Li et al. Mater. Sci. Eng. C, 2017, 74:434~442.
-
[109]
M Ornatska, E Sharpe, D Andreescu et al. Anal. Chem., 2011, 83:4273~4280.
-
[110]
Y Li, X He, J J Yin et al. Angew. Chem. Int. Ed., 2015, 54:1832~1835.
-
[111]
J S Mu, Y Wang, M Zhao. Chem. Commun., 2012, 48:2540~2542.
-
[112]
W Yang, J Hao, Z Zhang et al. New J. Chem., 2015, 39(11):8802~8806.
-
[113]
J Dong, L Song, J J Yin et al. ACS. Appl. Mater. Int., 2014, 6:1959~1970.
-
[114]
J S Mu, L Zhang, M Zhao et al. ACS. Appl. Mater. Int., 2014, 6:7090~7098.
-
[115]
W Qin, L Su, C Yang et al. J. Agric. Food Chem., 2014, 62:5827~5834.
-
[116]
Y Dong, Y Chi, X Lin et al. Phys. Chem. Chem. Phys., 2011, 13:6319~6324.
-
[117]
Y Wang, X Zhang, Z Luo et al. Nanoscale,2014, 6:12340~12344.
-
[118]
W Li, B Chen, H Zhang et al. Biosens. Bioelectron., 2015, 66:251~258.
-
[119]
A Hu, Y H Liu, H H Deng et al. Biosens. Bioelectron., 2014, 61:374~378.
-
[120]
Q Liu, Q Y Jia, R R Zhu et al. Mater. Sci. Eng. C, 2014, 42:177~184.
-
[121]
W Shi, W B Shi, X D Zhang et al. Chem. Commun., 2011, 47:10785~10787.
-
[122]
X Liu, Q Wang, H H Zhao et al. Analyst, 2012, 137:4552~4558.
-
[1]
-

-


 下载:
下载:




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
