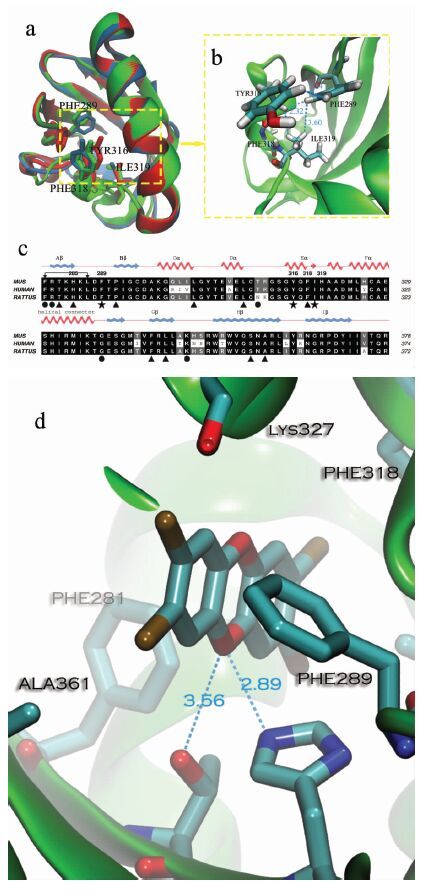
 图 1
(a)AhR LBD结构域构象(绿色:小鼠源;黄色:大鼠源;蓝色:人源)(b)鼠源AhR Phe289,Tyr316,Phe318,Ile319的空间构象(c)不同物种AhR LBD结构域序列比对 (d) mAhR与TCDD的结合模式
Figure 1.
(a)Conformations of AhR LBD domain (green: Mus;yellow: Rat; blue: Human);(b) Conformations of mAhR Phe289,Tyr316,Phe318 and Ile319 residues; (c)Sequence alignment in different species; (d) Representative binding modes of the mAhR and TCDD
图 1
(a)AhR LBD结构域构象(绿色:小鼠源;黄色:大鼠源;蓝色:人源)(b)鼠源AhR Phe289,Tyr316,Phe318,Ile319的空间构象(c)不同物种AhR LBD结构域序列比对 (d) mAhR与TCDD的结合模式
Figure 1.
(a)Conformations of AhR LBD domain (green: Mus;yellow: Rat; blue: Human);(b) Conformations of mAhR Phe289,Tyr316,Phe318 and Ile319 residues; (c)Sequence alignment in different species; (d) Representative binding modes of the mAhR and TCDD
引用本文:
康文渊, 徐锡明, 郭建秀, 田菲菲. 分子动力学模拟残基突变对芳香烃受体配体结合区的影响[J]. 化学通报,
2017, 80(2): 179-184, 207.

Citation: Kang Wenyuan, Xu Ximing, Guo Jianxiu, Tian Feifei. Molecular Dynamics Studies on the Structure of Aryl Hydrocarbon Receptor Fragment Affected by Site-Directed Mutagenesis[J]. Chemistry, 2017, 80(2): 179-184, 207.
Citation: Kang Wenyuan, Xu Ximing, Guo Jianxiu, Tian Feifei. Molecular Dynamics Studies on the Structure of Aryl Hydrocarbon Receptor Fragment Affected by Site-Directed Mutagenesis[J]. Chemistry, 2017, 80(2): 179-184, 207.
分子动力学模拟残基突变对芳香烃受体配体结合区的影响
摘要:
芳香烃受体(Aryl hydrocarbon receptor,AhR)属于配体依赖性的转录因子蛋白。本文通过对AhR配体结合区域(Ligand binding domain,LBD)的结构功能及物种特异性分析,发现在其结合腔口有一些关键残基可能起到“门控”作用,进一步将野生型(WT)和3个突变模型(Phe289Ala、Tyr316Ala、Ile319Ala)进行分子动力学模拟,从蛋白稳定性、蛋白结构变化、蛋白结合腔变化及蛋白和配体结合能力4个方面分析3个残基的门控作用。研究发现,Phe289、Tyr316、Ile319氨基酸残基通过形成疏水作用为AhR LBD起到“门控”作用;而将这些氨基酸分别突变后,其蛋白稳定性降低,整体运动性增加,配体亲和力减弱,其中Tyr316、Ile319对腔内体积影响较大,Phe289使腔内环境稳定性降低。本研究可为基于芳香烃受体的药物设计提供相关理论指导。
English
Molecular Dynamics Studies on the Structure of Aryl Hydrocarbon Receptor Fragment Affected by Site-Directed Mutagenesis
Abstract:
The Aryl hydrocarbon receptor (AhR), a ligand-actived nuclear transcription factor, regulates the expressions of a diverse group of genes when small molecules go into the pocket of its single ligand binding domain(LBD). In this study, we found Phe289, Tyr316 and Ile319 play "gatekeeper" roles via comparing genus-species specific and structure analysis. The four mutation models (WT, Phe289Ala, Tyr316Ala, Ile319Ala) were performed using the Amber99sb force field of GROMACS software package. The complex stability, protein conformational change, active site volume and binding free energy of the binary complex were analyzed by "gatekeeper" residues function. The results indicated that Phe289, Tyr316 and Ile319 play "gatekeeper" roles through hydrophobic interaction. The whole structure analyses demonstrate that mutagenesis comes from the pockets, which will destabilize cavity environment, causing obvious fluctuations and leading to low ligand-affinity and ligand escape. These simulation results interpret some experimental phenomena and provide further structural information that are useful in drug design based on aryl hydrocarbon receptor.
-
Key words:
- Molecular simulation
- / Molecular dynamics
- / Mutation
- / Aryl hydrocarbon receptor
-
芳香烃受体(AhR)属于碱性螺旋-环-螺旋(Basic helix-loop-helix,bHLH)转录因子超家族中的PAS(Per-Arnt-Sim)亚家族,可通过与一些环境污染物结合激活下游多种基因表达,进而介导致癌、致畸和生物转化等作用[1],这些环境污染物包括有毒的卤代芳香烃类(HAHs),多环芳香烃类(PAHs)及其衍生物[2],如多氯代二苯并-对-二噁英(PCDDs),二苯并呋喃(PCDFs),四氯二苯并-p-二噁英(TCDD)等。在非环境污染物作为配体时AhR则扮演不同的生理学角色,例如,影响细胞生长、分化和凋亡,调控自身免疫、炎症以及癌症进程[3]。
TCDD是具有致癌性的环境污染物,化学工作者意外暴露在化学试剂下导致氯痤疮暴发而使得TCDD被发现,因其特殊毒性和扩散性使得人们开始关注该化合物对人类健康可能造成的危害,TCDD对人体的毒害作用主要通过AhR介导。在众多小分子配体中,TCDD因其发现最早且具有与AhR的高亲和力而成为典型的和研究最多的AhR配体[4]。TCDD是通过AhR配体结合区(Ligand binding domain,LBD)与AhR结合,进而调节AhR及其下游信号通路的变化,发挥其毒性作用[5~7],因而对AhR LBD的研究就显得尤为重要,但目前该结构域未被解析[8]。
现在AhR LBD的研究很多,但仍有一些问题没被解释清楚:(1) AhR LBD突变引起配体活性降低的分子机制并不清楚;(2) 尚不明确AhR LBD中控制配体、水、离子进出的“门控”残基。Soshilov等[9]发现AhR的Phe289、Ile319突变成Ala后,将导致TCDD活性大幅度下降。他们[10, 11]将Tyr316、Ile319突变成Ala后,使得AhR对TCDD的结合活性降低超过50%;Goryo等[12]将Ile319突变成Ala后也得到相似的实验结果。根据这些实验现象,本文主要选择门控残基Phe289、Tyr316、Ile319突变成Ala进行研究,应用分子动力学模拟,从蛋白稳定性、蛋白结构变化、蛋白结合腔变化及蛋白的配体结合能力4个方面分析这3个残基,发现其“门控”作用,进而为针对AhR LBD结构靶点的药物设计、筛选和开发应用提供重要的理论依据。
1 实验部分
1.1 结构准备
人源、鼠源、大鼠源蛋白质序列结构来源于GenBank(ID:AAH70080.1,AAK13443.1,AAC35169.1) ,带有THS配体的HIF-2α PAS B晶体结构(PDB ID:3F1O[13],3H7W[14],3H82[14]),TCDD三维结构来源于NCBI数据库。
序列比对使用ClustalW 2.0[15],采用Modeller 9.16[16]用Motto[11]等提出的方法进行同源建模。模型采用基于配体的多模板建模,进行loop环优化,能量最小化,每个步骤生成100个模型,ERRAT2评分最高的模拟被选择。突变模型使用PyMOL相关功能完成Phe289、Tyr316、Ile319突变成Ala。
活性位点方法参照之前研究[17],分子对接采用AutoDock Vina刚性对接,盒子大小设置恰好能包括AhR结合腔,每次对接生成10个构象,评分最高构象被选择。蛋白稳定性使用FoldX套件[18]中的Repair object和Stability两个模块完成。
1.2 分子动力学模拟
对鼠源的AhR野生型(WT)和3个突变型(Phe289Ala,Tyr316Ala,Ile319Ala)模型使用GROMACS分子动力学模拟软件(版本5.0.6) Amber99sb力场模拟[19]。运用十二立方体盒子的Tip3p水模型,每个系统的蛋白与盒子距离设置为1.0nm;TCDD使用Amber推荐的GAFF力场,具体方法为由Gaussian09计算RESP,基组选择HF/6-31G,具体参数为SCF=tight Test Pop=MK iop(6/33=2) iop(6/42=6) iop(6/50=1) ,后通过Amber中Antechamber工具以及ACPYPE项目转化为TCDD拓扑文件。
1.3 数据分析方法
碳链的骨架均方根偏差(RMSD)、C-αRMSF、Gyrate、DSSP、g_helix均使用Gromacs自带分析工具完成。结合自由能分析使用基于MM/PBSA算法的g_mmpbsa工具[20]。口袋构象的静态变化和动态变化使用Epock[21]项目来计算。
结构比对图运用Aline[22]工具,2D绘图采用Veusz,三维结构图使用VMD[23]完成。
2 结果与讨论
2.1 AhR LBD结合模式和突变稳定性分析
AhR LBD与已解析晶体结构最大相似度只有27%,而AhR LBD所属的PAS结构域结构具有高度保守性,且PAS家族HIF2α蛋白的两个配体(THS-10,THS17) 也对AhR有亲和力[11],故小鼠源、大鼠源以及人源AhR LBD的模型生成使用含有配体的HIF2α蛋白结构作为模版(PDB ID:3F1O,3H7W,3H82) ,模型优化后评分较佳,说明模型质量可靠。将三个模型叠合(图 1(a))并进行序列比对分析(图 1(c))。
图 1
(a)AhR LBD结构域构象(绿色:小鼠源;黄色:大鼠源;蓝色:人源)(b)鼠源AhR Phe289,Tyr316,Phe318,Ile319的空间构象(c)不同物种AhR LBD结构域序列比对 (d) mAhR与TCDD的结合模式
Figure 1.
(a)Conformations of AhR LBD domain (green: Mus;yellow: Rat; blue: Human);(b) Conformations of mAhR Phe289,Tyr316,Phe318 and Ile319 residues; (c)Sequence alignment in different species; (d) Representative binding modes of the mAhR and TCDD
从图 1(a)可以发现,Phe289(位于A-B-loop环结构上)、Tyr316、Phe318和Ile319(位于E-alpha螺旋上)4个氨基酸残基均位于AhR LBD的腔口,构象无较大变化,其残基侧链均位于腔的内部,且它们在不同种属中具有高度保守性(图 1(c))。进一步对loop环和alpha螺旋分析(图 1(b))发现,Phe289、Tyr316、Ile319残基侧链较长,彼此间靠疏水作用相互联系,成为AhR LBD结合腔的屏障,维持结合腔的稳定。Tyr316、Phe289苯环和Ile319侧链甲基距离小于3.6Å,使得水分子无法进入,Phe318侧链朝Ile319相反一侧,并未参与其中,提示3个氨基酸残基Phe289、Tyr316和Ile319形成的疏水作用起到“门控”作用,能够“开关”AhR LBD结合腔,控制着配体、离子和水分子的进出。
此外,将配体TCDD对接入腔内,分析AhR与配体的结合模式发现,TCDD与AhR上氨基酸残基Phe281、Phe318等形成疏水作用紧密结合(图 1(d)),使得TCDD能够牢牢卡在结合腔中;而Thr283侧链羟基、His285侧链咪唑环与TCDD上氧原子形成氢键进一步稳定体系;Phe289、Tyr316、Ile319在较远端为TCDD提供疏水屏障。
为了验证上述分析,将Phe289、Tyr316、Ile319三个“门控”位点分别突变成Ala,进行分子动力学研究,Phe318不具备“门控”残基性质,所以未纳入分析。通过基于骨架原子的RMSD分析是否趋于平衡(图 2(a))来评价模拟50ns后模型的质量,结果表明,野生型和Tyr316Ala突变模型AhR LBD的RMSD稳定在0.15nm左右,但前者10ns时发生一个轻微构象波折,后者则是非常迅速的平衡;Ile319Ala突变模型30ns后趋于平衡,平衡后RMSD值分别稳定在0.20nm和0.22nm;Phe289Ala在50ns内一直未平衡,且波动较大,对其延长50ns模拟(图 2(b)),发现约70ns后趋于平衡,平衡后RMSD值约为0.26nm,提示Tyr316Ala以外的突变模型相比野生模型具有更大的结构变化,其中,Phe289Ala突变模型尤为明显。随后进行蛋白回转半径分析,该程序一定程度上能说明蛋白的形状变化,分析表明,蛋白形状变化均趋于稳定,未出现较大的波动,但4种突变模型相比野生型回旋半径增大,提示AhR LBD结构在突变后形状发生了改变。
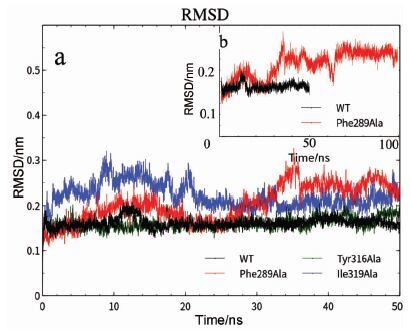 图 2
4种模型蛋白模拟的RMSD曲线
Figure 2.
Backbone root mean square deviations (RMSD) of the four models as a function of time
图 2
4种模型蛋白模拟的RMSD曲线
Figure 2.
Backbone root mean square deviations (RMSD) of the four models as a function of time
运用FoldX[16]结合Yasara项目对模拟平衡后结构进行稳定性分析,检测突变是否会对蛋白稳定造成影响。从表 1所列结果可知,氨基酸突变后蛋白整体能量升高,稳定性降低,说明这3个突变残基在维持蛋白稳定性中起到重要作用,猜测可能涉及蛋白结构的变化、配体的亲和力、腔内环境稳态、下游蛋白的激活等因素,为此进一步分析突变模型这些因素的变化。
 表 1
蛋白稳定性分析
Table 1.
Protein stability in FoldX
表 1
蛋白稳定性分析
Table 1.
Protein stability in FoldX
2.2 残基突变对蛋白结构变化分析
C-α碳原子的均方根波动(Root mean square fluctuations,RMSF)可用来表征蛋白残基突变后的柔性变化情况(图 3) 。将Phe289突变成Ala后,289、316、332、350及附近氨基酸残基柔性变大;Tyr316突变成Ala后,316、350及附近氨基酸残基柔性变大;Ile319突变成Ala后,316、332、350及附近氨基酸残基柔性变大。分析原因发现,289位点上Phe突变成Ala后,使得Phe289、Tyr316、Ile319之间疏水作用瓦解,造成了289、316、332附近氨基酸残基不稳定。350周围残基位于结合腔内侧(图 1(a)),摆动性增加原因可能是“门控”残基形成的疏水联系打破,从而导致疏水腔内环境暴露而变得不稳定。316位点上Tyr突变为Ala,使得316位点和350位点摆动陡增,说明腔口的疏水联系被破坏,原本稳定的腔口环境变得不稳定;但Phe289与Ile319产生的疏水联系并未瓦解,两者摆动性也没有增加。319位点上Ile突变成Ala并未造成289残基周围柔性变大,原因是与Phe289之间疏水联系较为薄弱(3.6Å),Ile319更多为Tyr316提供的是屏障作用,350附近氨基酸残基柔性并未增加提示口袋体系相对稳定。
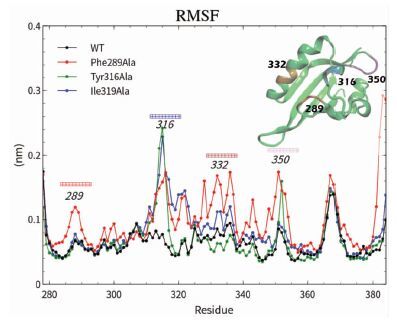 图 3
4种模型蛋白模拟Cα的RMSF曲线
Figure 3.
Root mean square fluctuations (RMSF) of residues in the protein during dynamics simulations
图 3
4种模型蛋白模拟Cα的RMSF曲线
Figure 3.
Root mean square fluctuations (RMSF) of residues in the protein during dynamics simulations
进一步对模拟的初始构象和平衡后平均构象绘制“刺猬图”(Porcupine plot),发现运动主要发生在A-B-loop环(289位点附近)、E-alpha螺旋(316位点附近)和H-I-loop环(350位点)区域(图 4) 。具体来说,Phe289Ala中E-alpha螺旋和A-B-loop环之间距离增大,H-I-loop环凹入腔内;Tyr316Ala中虽然A-B-loop环区域变化不明显,但E-alpha螺旋大幅度波动;Ile319Ala中E-alpha螺旋与A-B-loop环之间距离减小。以上结果提示,Phe289Ala突变导致结合腔腔口之间疏水作用丧失,从而A-B-loop环与E-alpha螺旋联系减弱,两者分开。Tyr316Ala突变导致E-alpha摆动增加,这是由于316位点贡献在E-alpha螺旋上的疏水作用消失,使得腔口该区域受到水分子的冲击变得稳定性降低。A-B-loop环影响较小可能与Phe289Ala和Ile319Ala之间疏水作用仍然存在有关。Ile319Ala突变使得E-alpha螺旋与A-B-loop环之间空间距离拉近,可能是319位点贡献的疏水作用力被Phe289与Tyr316之间更为紧密的联系所弥补,说明Phe289突变较Ile319突变对于“门控”残基疏水作用联系的影响更为致命。
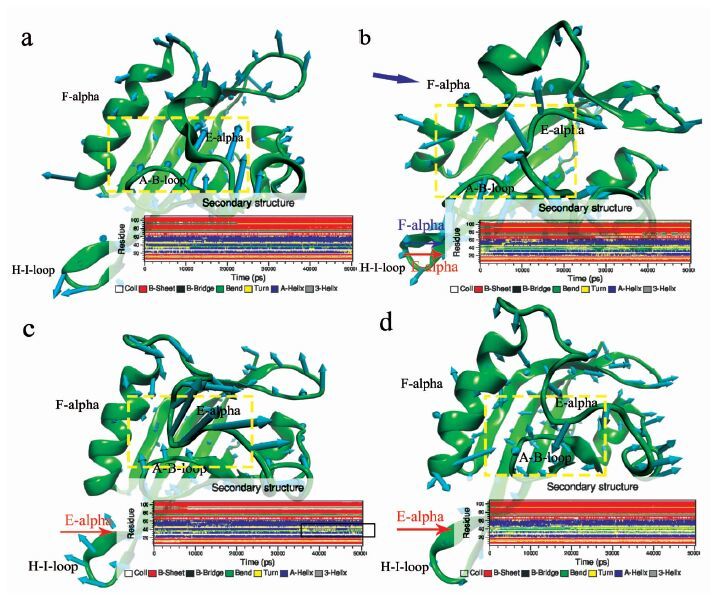 图 4
4种模型的蛋白运动刺猬图和二级结构随时间的变化图 (a)野生型;(b)Phe289Ala;(c)Tyr316Ala;(d)Ile319Ala
Figure 4.
Porcupine plot of the first eigenvector and secondary structure changes of the AhR LBD domain residues as a function of time (a) WT; (b)Phe289Ala; (c)Tyr316Ala; (d)Ile319Ala
图 4
4种模型的蛋白运动刺猬图和二级结构随时间的变化图 (a)野生型;(b)Phe289Ala;(c)Tyr316Ala;(d)Ile319Ala
Figure 4.
Porcupine plot of the first eigenvector and secondary structure changes of the AhR LBD domain residues as a function of time (a) WT; (b)Phe289Ala; (c)Tyr316Ala; (d)Ile319Ala
采用DSSP分析AhR蛋白结构随时间的变化(图 4) ,发现3个突变模型中alpha螺旋占有率大幅度降低,Phe289Ala、Ile319Ala突变的E-alpha螺旋变为无规卷曲或弯曲,Tyr316Ala的则为螺旋弯曲混合交替,说明Phe289、Tyr316、Ile319三者疏水作用对维系E-alpha螺旋结构具有重要的作用。
Phe289Ala突变模拟初始构象显示F-alpha螺旋变形(图 4(b)),但在模拟过程中并未发现F-alpha螺旋性质明显的变化,可能仅是动态变化中不同的形态,并未有统计学意义上的差异,这也体现了分子动力学模拟捕捉准确信息的优越性。
2.3 残基突变结合腔变化分析
观察Phe289、Tyr316、Ile319氨基酸突变前后腔内环境的变化,采用Epock项目计算野生型和突变型模型结构的口袋动态变化,并查看氨基酸残基对腔体积的贡献程度。
通过口袋体积随时间的变化图(图 5) 发现,AhR野生型在10ns后口袋趋于非常稳定,平均口袋体积约410Å3;Phe289Ala突变的口袋体积增加,约为450Å3;Tyr316Ala和Ile319Ala突变后口袋体积降低,平均为360Å3,突变模型结合腔口袋体积变化程度均大于野生型。
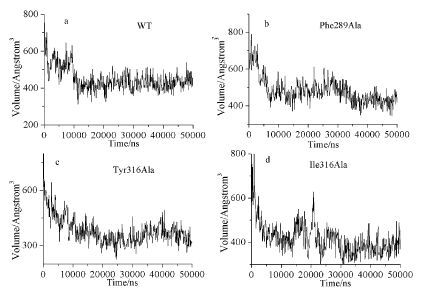 图 5
4种不同模型的口袋随着时间的变化
Figure 5.
Volumes of the active pockets of the four models as a function of time
图 5
4种不同模型的口袋随着时间的变化
Figure 5.
Volumes of the active pockets of the four models as a function of time
结合腔变化分析表明,三个位点的突变对结合口袋的体积与形态造成影响,Phe289Ala突变导致Tyr316因为疏水作用联系消失而暴露在腔内,使得Phe289Ala体积轻微增加(30Å3),Tyr316Ala和Ile319Ala突变则导致A-B-loop环与E-alpha螺旋上疏水氨基酸残基为了降低疏水作用的影响而更彼此“靠近”以缓解水分子的“侵袭”,同时腔内疏水氨基酸残基为了弥补突变造成的突变位点疏水贡献的空缺而“压缩”,使得腔变得更浅(图 6) ,说明Phe289、Tyr316和Ile319对与维系腔内体积起到关键作用。
 图 6
结合口袋分析(a)野生型;(b)Tyr316Ala;(c)Ile319Ala
Figure 6.
Active pocket analysis of (a)WT,(b)Tyr316Ala and (c)Ile319Ala
图 6
结合口袋分析(a)野生型;(b)Tyr316Ala;(c)Ile319Ala
Figure 6.
Active pocket analysis of (a)WT,(b)Tyr316Ala and (c)Ile319Ala
2.4 残基突变与配体结合自由能分析
使用MM/PBSA方法分析TCDD配体与突变蛋白的结合能力,计算真空潜在自由能、极性溶剂化能和非极性溶剂化能。
由于真实环境并非水模型那么简单,且结合能的计算并未包含难以捕捉的熵变,故仅对线性关系进行分析。表 2数据表明,Tyr316Ala、Ile319Ala两者与配体的结合能低于野生型AhR与TCDD配体的结合能,但Phe289Ala的结合自由能并未发现亲和力降低,与野生型数据相差不大。Phe289Ala突变模型在蛋白结构变化分析中结构变化差异较大,猜测可能是TCDD出现了逃逸现象而导致结合自由能数据与实验数据不同。根据轨迹构象,均匀地提取20个位点来查看TCDD的运动轨迹(图 7) 。野生型模拟后TCDD位置并未明显移动,而Phe289Ala突变型AhR中TCDD移动较为明显,且有较大的翻转空间,而翻转普遍被认为是亲和力差的体现[24]。原因在于,Phe289Ala突变造成A-B-loop环(Phe289氨基酸残基附近)与E-alpha螺旋之间的稳态被破坏,而“裂开”给TCDD提供了更多的活动空间。
表 2
AhR蛋白模型结合自由能
Table 2.
Partial binding energy of ligands with AhR protein
Electrostatic energy (kJ/mol) -3.188+/-2.773 -4.790+/-1.520 -5.350+/-3.777 -1.704+/-2.819 Polar salvation energy (kJ/mol) 66.208+/-6.865 63.962+/-6.243 64.516+/-8.833 64.573+/-3.824 SASA (kJ/mol) -15.243+/-0.612 -15.272+/-0.607 -10.954+/-0.795 -9.024+/-0.666 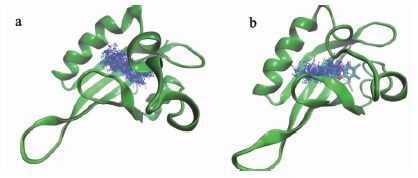 图 7
模拟过程中TCDD运动轨迹(a) 野生型; (b) Phe289Ala
Figure 7.
Ligand motions in (a)WT and (b)Phe289Ala simulation ensembles
图 7
模拟过程中TCDD运动轨迹(a) 野生型; (b) Phe289Ala
Figure 7.
Ligand motions in (a)WT and (b)Phe289Ala simulation ensembles
将模拟后的蛋白进行再对接,构象位置发生了很大的变化,一定程度上TCDD进行了翻转,对接评分也有所降低。以上证据均表明TCDD逃逸的直接原因是A-B-loop环与E-alpha螺旋直接的联系被破坏,表明“门控”残基疏水作用在维系配体腔内稳定起到重要作用。
3 结论
越来越多研究证实芳香烃受体在炎症、肿瘤等疾病发生中扮演者重要的角色,是一个潜在的药物靶点,而“门控”残基往往作为药物设计考虑的关键因素[25],本文选取结合腔口的Phe289、Tyr316、Ile319三个代表性氨基酸残基结合位点研究突变对AhR LBD结构造成的影响。运用分子动力学模拟的方法,从突变蛋白稳定性、二级结构变化、配体结合自由能变化和结合口袋变化4个方面分析,研究表明Phe289、Tyr316、Ile319三个疏水性氨基酸残基形成的疏水作用维系A-B-loop环与E-alpha螺旋的联系,影响蛋白结构变化、配体亲和力及腔内环境稳态。研究表明,Phe289、Tyr316、Ile319三个氨基酸残基对于AhR蛋白起到“门控”作用,对配体和腔内环境稳定起到关键作用。本研究结果为深入认识配体对AhR蛋白的作用机制以及以AhR蛋白为靶点的药物设计、筛选和产品开发提供了重要理论依据。
-
-
[1]
B W Smith, S S Rozelle, A Leung et al. Blood, 2013, 122:376~385. doi: 10.1182/blood-2012-11-466722
-
[2]
P Y Chan-Hui, K Stephens, R A Warnock et al. Clin. Immunol., 2004, 111:162~174. doi: 10.1016/j.clim.2003.12.015
-
[3]
A J Parks, M P Pollastri, M E Hahn et al. Mol. Pharmacol., 2014, 86:593~608. doi: 10.1124/mol.114.093369
-
[4]
B Stockinger, P Di Meglio, M Gialitakis et al. Ann. Rev. Immunol., 2014, 32:403~432. doi: 10.1146/annurev-immunol-032713-120245
-
[5]
J V Schmidt, C A Bradfield. Ann. Rev. Cell. Dev. Biol., 1996, 12:55~89. doi: 10.1146/annurev.cellbio.12.1.55
-
[6]
P D Kottaridis, R E Gale, M E Frew et al. Blood, 2001, 98:1752~1759. doi: 10.1182/blood.V98.6.1752
-
[7]
R J Kewley, M L Whitelaw, A Chapman-Smith. Int. J. Biochem. Cell Biol., 2004, 36:189~204. doi: 10.1016/S1357-2725(03)00211-5
-
[8]
R Farmahin, G E Manning, D Crump et al. Toxicol. Sci., 2013, 131:139~152. doi: 10.1093/toxsci/kfs259
-
[9]
A A Soshilov, M S Denison. Mol. Cell. Biol., 2014, 34:1707~1719. doi: 10.1128/MCB.01183-13
-
[10]
A Pandini, A A Soshilov, Y Song et al. Biochemistry, 2009, 48:5972~5983. doi: 10.1021/bi900259z
-
[11]
I Motto, A Bordogna, A A Soshilov et al. J. Chem. Inf. Model, 2011, 51:2868~2881. doi: 10.1021/ci2001617
-
[12]
K Goryo, A Suzuki, C A del Carpio et al. Biochem. Biophys. Res. Commun., 2007, 354:396~402. doi: 10.1016/j.bbrc.2006.12.227
-
[13]
J Key, T H Scheuermann, P C Anderson et al. J. Am. Chem. Soc., 2009, 131:17647~17654. doi: 10.1021/ja9073062
-
[14]
M Bajpai. IDrugs, 2009, 12:174~185.
-
[15]
M A Larkin, G Blackshields, N P Brown et al. Bioinformatics, 2007, 23:2947~2948. doi: 10.1093/bioinformatics/btm404
-
[16]
M A Marti-Renom, A C Stuart, A Fiser et al. Ann. Rev. Biophys. Biomol. Struct., 2000, 29:291~325. doi: 10.1146/annurev.biophys.29.1.291
-
[17]
康文渊, 丁若凡, 范倩等. 化学通报, 2015, 78:944~944. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20150428003&flag=1
-
[18]
N Tokuriki, F Stricher, J Schymkowitz et al. J. Mol. Biol., 2007, 369:1318~1332. doi: 10.1016/j.jmb.2007.03.069
-
[19]
J A Maier, C Martinez, K Kasavajhala et al. J. Chem. Theory. Comput., 2015, 11:3696~3713. doi: 10.1021/acs.jctc.5b00255
-
[20]
R Nuti, M Gargaro, D Matino et al. J. Chem. Inf. Model, 2014, 54:3373~3383. doi: 10.1021/ci5005459
-
[21]
B Laurent, M Chavent, T Cragnolini et al. Bioinformatics, 2015, 31:1478~1480. doi: 10.1093/bioinformatics/btu822
-
[22]
C S Bond, A W Schuttelkopf. Acta Crystallogr., Sect D:Biol. Crystallogr., 2009, 65:510~512. doi: 10.1107/S0907444909007835
-
[23]
W Humphrey, A Dalke, K Schulten. J. Mol. Graphics, 1996, 14:33~38. doi: 10.1016/0263-7855(96)00018-5
-
[24]
N Widderich, M Pittelkow, A Hoppner et al. J. Mol. Biol., 2014, 426:586~600. doi: 10.1016/j.jmb.2013.10.028
-
[25]
A Unzue, H Zhao, G Lolli et al. J. Med. Chem., 2016, 59:3087~3097. doi: 10.1021/acs.jmedchem.5b01757
-
[1]
-

图 1 (a)AhR LBD结构域构象(绿色:小鼠源;黄色:大鼠源;蓝色:人源)(b)鼠源AhR Phe289,Tyr316,Phe318,Ile319的空间构象(c)不同物种AhR LBD结构域序列比对 (d) mAhR与TCDD的结合模式
Figure 1 (a)Conformations of AhR LBD domain (green: Mus;yellow: Rat; blue: Human);(b) Conformations of mAhR Phe289,Tyr316,Phe318 and Ile319 residues; (c)Sequence alignment in different species; (d) Representative binding modes of the mAhR and TCDD

图 2 4种模型蛋白模拟的RMSD曲线
Figure 2 Backbone root mean square deviations (RMSD) of the four models as a function of time

图 3 4种模型蛋白模拟Cα的RMSF曲线
Figure 3 Root mean square fluctuations (RMSF) of residues in the protein during dynamics simulations

图 4 4种模型的蛋白运动刺猬图和二级结构随时间的变化图 (a)野生型;(b)Phe289Ala;(c)Tyr316Ala;(d)Ile319Ala
Figure 4 Porcupine plot of the first eigenvector and secondary structure changes of the AhR LBD domain residues as a function of time (a) WT; (b)Phe289Ala; (c)Tyr316Ala; (d)Ile319Ala

图 5 4种不同模型的口袋随着时间的变化
Figure 5 Volumes of the active pockets of the four models as a function of time

图 6 结合口袋分析(a)野生型;(b)Tyr316Ala;(c)Ile319Ala
Figure 6 Active pocket analysis of (a)WT,(b)Tyr316Ala and (c)Ile319Ala

图 7 模拟过程中TCDD运动轨迹(a) 野生型; (b) Phe289Ala
Figure 7 Ligand motions in (a)WT and (b)Phe289Ala simulation ensembles
表 1 蛋白稳定性分析
Table 1. Protein stability in FoldX
 下载: 导出CSV
下载: 导出CSV
表 2 AhR蛋白模型结合自由能
Table 2. Partial binding energy of ligands with AhR protein
Electrostatic energy (kJ/mol) -3.188+/-2.773 -4.790+/-1.520 -5.350+/-3.777 -1.704+/-2.819 Polar salvation energy (kJ/mol) 66.208+/-6.865 63.962+/-6.243 64.516+/-8.833 64.573+/-3.824 SASA (kJ/mol) -15.243+/-0.612 -15.272+/-0.607 -10.954+/-0.795 -9.024+/-0.666
下载: 导出CSV
-








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
