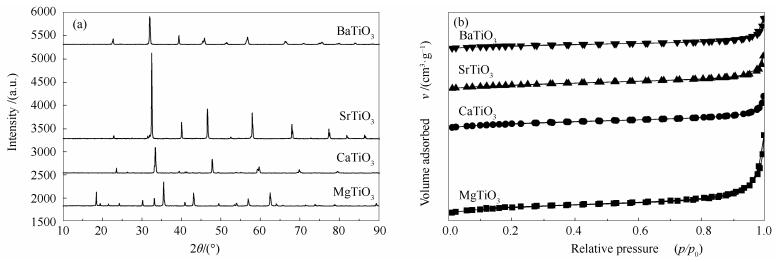
图 1.
钙钛矿载体的XRD谱图(a)和N2吸附等温线(b)
Figure 1.
XRD patterns (a) and N2 sorption isotherms (b) of various MTiO3 perovskite supports

煤炭、石油和天然气是三大化石能源。随着经济社会的发展,世界能源消费结构发生了深刻变化,以煤炭和石油消费为主的能源结构已经难以满足人类可持续发展的需求。近年来,随着页岩气和可燃冰开采技术的突破,清洁高效的天然气利用技术再次受到了广泛关注[1, 2]。甲烷是常规天然气和页岩气的主要成分, 甲烷在化工中的应用主要有两种方式:直接转化和间接转化[3, 4]。
甲烷直接转化是指将甲烷直接转化为乙烯、芳烃及醇类等重要的化学工业品[5-7],然而,由于甲烷分子结构的高度稳定而难以活化,直接转化时条件苛刻,甲烷转化率和产物选择性均不甚理想。目前,甲烷的直接转化仍然停留在基础研究阶段,短期内难以实现大规模的工业应用。甲烷的间接转化是指先将甲烷通过重整手段转化为合成气(CO和H2),然后将合成气在适当的催化剂上高效地转化为所需的化学品和燃料,是具有应用前景的甲烷转化途径。
甲烷重整制合成气的途径主要包括甲烷水蒸气重整(SRM)、甲烷部分氧化重整(POM)、甲烷二氧化碳重整(DRM)。其中,SRM是研究最早也是工业上应用最为广泛的制备合成气的方法[8]。但是该方法耗能大、反应器制作成本高、较高的反应温度易导致催化剂失活[9, 10]。而且,该反应途径制得的合成气中H2/CO(体积比)≈3,不利于后续的甲醇和费托合成[11]。POM过程放热温和、能耗低、产物中H2/CO(体积比)≈2,适用于后续的费托合成和甲醇合成[12, 13],但存在着空分氧成本高、CH4和O2混合有较高的爆炸风险、催化剂床层易飞温等问题。CH4和CO2是两种典型的温室气体,又是重要的含碳资源。DRM将CH4和CO2作为碳源和氧源,在一定条件下转化成合成气,不仅具有良好的环境效益,而且该反应制备的合成气中H2/CO(体积比)≈1,有利于下游的羰基合成及费托合成[14]。因此,DRM被认为是甲烷间接利用的较佳途径。
DRM是一个强吸热反应,而且CH4和CO2分子都比较难以活化,因此,为了实现较高的原料转化率,DRM反应一般在较高温度下(>700℃)进行[15]。积炭是制约甲烷二氧化碳重整工业化的重要瓶颈之一[16]。虽然贵金属催化剂具有良好的催化活性和抗积炭性能,但是贵金属高昂的价格限制了其大规模应用。廉价的Ni基催化剂在该反应中可以表现出与贵金属相当的活性,但是其抗积炭性能和稳定性有待提高。因此,如何提高Ni基催化剂的抗积炭性能、延长催化剂的寿命是该反应的重点研究方向之一[17]。
DRM反应需要催化剂具有甲烷裂解和二氧化碳吸附活化两种活性位。甲烷裂解主要发生于活性金属表面,是积炭的主要来源;二氧化碳的吸附活化则与载体性质息息相关,决定了催化剂的消炭能力[17, 18]。因此,负载型金属催化剂中载体的性质对催化剂抗积炭性能有重要的影响:第一为载体的织构性质。具有丰富的孔道结构以及大比表面积的载体可以促进活性组分的分散。活性组分的颗粒越小其抗积炭性能越强[19]。Arbag等[20]将Ni/MCM-41催化剂用于DRM反应中,结果表明由于载体具有高的比表面积,活性组分得以高度分散,所以催化剂具有较好的DRM反应性能。第二为载体的酸碱性。载体的酸碱性会影响CO2的吸附活化,进而影响催化剂的性能。载体的碱性越强越容易吸附、活化CO2,利于积炭的快速消除[21, 22],从而保持催化剂的稳定性。Wang等[23]将Al2O3、SiO2、MgO、La2O3以及活性炭等载体制备的催化剂用于DRM反应中,发现MgO和La2O3由于具有较高的碱性,以其为载体的催化剂表现出良好的抗积炭性能。对于使用SiO2等惰性载体的催化剂,在反应过程中CH4和CO2的活化均发生在活性金属表面,此时,CO2的吸附活化以及积炭的氧化均受到了限制,较易产生积炭[24]。第三为载体表面氧物种的作用。Zhang等[17, 25]注意到ZrO2表面的吸附氧物种对于CO2的活化有一定的贡献,且通过掺杂稀土元素可以调节催化剂表面吸附氧物种的含量。第四为载体与活性组分之间的相互作用。Yan等[26]研究发现,Ni/TiO2催化剂中金属和载体间相互作用较强,且TiO2载体中存在活泼的氧物种,Ni/TiO2催化剂在DRM反应中有较好的活性和抗积炭性能。Akri等[27]研究发现,Ce掺杂HAP会导致强的金属-载体相互作用,使Ni单原子保持稳定,可以避免积炭,使催化剂表现出了较高活性和稳定性。
具有ABO3结构的钙钛矿型氧化物的A位元素一般是稀土或碱土元素,B位元素为过渡金属元素。该类氧化物具有以下主要特征:第一,结构稳定,耐高温;第二,晶格结构对离子半径的容纳范围较大,A、B位离子可以被部分或全部替换而不会影响结构;第三,具有良好的氧迁移能力。Hayakawa等[28]采用固相结晶法制备了以钙钛矿型复合氧化物为载体的负载型Ni基催化剂,发现钙钛矿载体可以高度分散Ni颗粒,从而提高催化剂的抗积炭性能。Li等[29]考察了Ni/BaTiO3和Ni/γ-Al2O3上催化剂DRM反应性能,结果表明,具有更强碱性的BaTiO3载体吸附活化CO2,移除积炭的能力更强,因此,Ni/BaTiO3催化剂有着更为优异的稳定性。
本研究采用MgTiO3、CaTiO3、SrTiO3、BaTiO3四种A位元素为不同碱土金属的钙钛矿型氧化物为载体,采用浸渍法制备了系列Ni基催化剂。通过XRD、H2-TPR、CO2-TPD、BET、TG、XPS等分析表征技术对反应前后的催化剂进行分析,并结合催化剂活性评价实验,探究了调变A位金属元素对钙钛矿型氧化物载体性质及其负载Ni催化剂的DRM反应行为的影响。
钛酸镁(MgTiO3, 99%)、钛酸钙(CaTiO3, 99.5%)、钛酸锶(SrTiO3, 99.5%)、钛酸钡(BaTiO3, 99.5%)购于Aladdin Industrial Corporation;六水合硝酸镍(Ni(NO3)2·6H2O,98.0%)购于国药集团化学试剂有限公司;无水乙醇(CH3CH2OH, 99.7%)购于天津市风船化学试剂科技有限公司。
采用浸渍法制备质量分数5%Ni/MTiO3(M=Mg、Ca、Sr、Ba)催化剂。按照化学计量比称取一定量的Ni(NO3)2·6H2O超声溶解于30mL无水乙醇中,将一定量的钙钛矿载体加入上述溶液,利用旋转蒸发仪将过量的乙醇蒸干。随后将获得的催化剂前驱体放入鼓风干燥箱内于110℃干燥12 h。干燥后的样品置于马弗炉内在700℃下空气气氛焙烧4h,最终得到实验所需催化剂。
催化剂的比表面积、孔体积等根据N2物理吸附法在Micromeritics Tristar 302自动吸附仪上进行测定。催化剂的比表面积根据BET (Brunauer-Emmet-Teller)方法进行计算。
催化剂的XRD谱图在Rigaku MiniFlexII射线衍射仪上测试。Cu靶射线,操作电压为40kV,电流为15mA,10°-90°扫描,扫描速率4(°)/min。
催化剂的H2-TPR测试在TP-5080多功能吸附仪上进行。首先,100mg催化剂置于反应管中部,并通入流量为30mL/min的Ar,升温至300℃,预处理30 min以除去催化剂表面杂质。而后降温至100℃,并将Ar切换为H2/Ar混合气(体积分数10%,30mL/min),观察并记录曲线。待基线平稳后,进行程序升温还原,以10℃/min的速率升温至700℃。H2的消耗由热导检测器(TCD)分析记录。
催化剂的CO2-TPD在TP-5080多功能吸附仪上进行,催化剂用量为100mg。测试时,样品首先在体积分数10%的H2/Ar混合气中预还原,温度控制在100min升温至700℃,并在该温度下保持60min。随后切换为N2吹扫并降温至50℃。体系内通入CO2,吸附30min。吸附完成后用N2吹扫30min,最后以10℃/min的速率升温至700℃,TCD记录CO2的脱附曲线。
XPS测定在Krato Axis Ultra DLD仪器上进行。以Al Kα(1486.6eV)为激光源,操作电压12kV,电流15mA。所有元素的电子结合能使用C 1s (284.6eV)进行校正。
TG在Rigaku TG-8120热重分析仪上进行。样品装填量10mg,在空气气氛下以20℃ /min的升温速率从室温升至850℃。
催化剂DRM反应性能评价在石英管反应器上进行。反应器内径为8mm,轴向中心装有外径为4mm的热电偶套管。将400mg颗粒粒径为20-40目的催化剂,用2000mg相同目数的石英砂稀释。稀释后的催化剂置于石英管中部,其余空间使用磁环填充。反应之前,催化剂在体积分数10%H2/Ar混合气氛下升温至700℃,升温时间为100min。待催化剂在700℃还原60min后,将还原气切换为CH4/CO2=1:1的反应原料气,进行反应,反应空速为GHSV=24000mL/(g·h)。反应出口气经室温冷阱冷却后进入色谱进行在线检测。
反应产物用气相色谱GC4000A进行检测分析(Ar为载气,填充柱型号为TDX-01色谱柱,热导检测器)。反应物转化率计算公式如下:
|
$ {\rm{C}}{{\rm{H}}_4}{\rm{conversion}}\left( \% \right) = \frac{{{V_{{\rm{C}}{{\rm{H}}_{\rm{4}}}{\rm{,in}}}} - {V_{{\rm{C}}{{\rm{H}}_{\rm{4}}}{\rm{,out}}}}}}{{{V_{{\rm{C}}{{\rm{H}}_{\rm{4}}}{\rm{,in}}}}}} \times 100\% $ |
(1) |
|
$ {\rm{C}}{{\rm{O}}_2}{\rm{conversion}}\left( \% \right) = \frac{{{V_{{\rm{C}}{{\rm{O}}_{\rm{2}}}{\rm{,in}}}} - {V_{{\rm{C}}{{\rm{O}}_{\rm{2}}}{\rm{,out}}}}}}{{{V_{{\rm{C}}{{\rm{O}}_{\rm{2}}}{\rm{,in}}}}}} \times 100\% $ |
(2) |
|
$ {{\text{H}}_{2}}/\text{CO }\left( \text{volume ratio} \right)=\frac{{{V}_{{{\text{H}}_{\text{2}}}\text{, out}}}}{{{V}_{\text{CO, out}}}} $ |
(3) |
式中,Vin为原料中气体流量,Vout为产物气体流量。
图 1(a)为购买的四种钙钛矿载体的XRD谱图。由图 1可知,四种钙钛矿的衍射峰高度有着明显差异。SrTiO3载体的衍射峰明显高于其他三种载体,且其衍射峰的半峰宽较其他三种载体小,采用Scherrer公式计算了四种钙钛矿型复合氧化物载体的晶粒粒径,结果见表 1。由表 1可知,SrTiO3载体的颗粒粒径比其他三种载体要大。
 下载:
导出CSV
下载:
导出CSV
| Sample | Surface area A/(m2·g-1) |
Pore volume v/(cm3·g-1) |
Crystalline sizea /nm |
Sample | Surface area A/(m2·g-1) |
Pore volume v/(cm3·g-1) |
| MgTiO3 | 4.98 | 0.0106 | 44.0 | Ni/MgTiO3 | 6.05 | 0.0190 |
| CaTiO3 | 3.65 | 0.0049 | 34.0 | Ni/CaTiO3 | 3.05 | 0.0065 |
| SrTiO3 | 3.53 | 0.0048 | 53.2 | Ni/SrTiO3 | 4.04 | 0.0070 |
| BaTiO3 | 2.56 | 0.0039 | 34.6 | Ni/BaTiO3 | 3.20 | 0.0067 |
| a: calculated by Scherrer equation according to the XRD results | ||||||
图 1(b)为四种钙钛矿载体的N2吸附等温线图。由图 1(b)可知,四种载体的吸附等温线均为Ⅳ型等温线,而且都没有明显的回滞环,说明四种载体的孔道结构都不甚发达。表 1的数据证明了四种钙钛矿型复合氧化物载体的比表面积较小。但是从图 1中也可以看出,MgTiO3载体的饱和吸附平台的相对压力较低,说明其具有较大的孔体积,这一点与表 1的数据相吻合。此外,MgTiO3相对大的比表面积也暗示其能够较其他三种载体更好地分散引入表面Ni物种。
图 2(a)是制备的新鲜Ni催化剂的XRD谱图。由图 2(a)可知,活性组分的负载和焙烧过程并未对催化剂的晶型结构造成太大影响。根据文献报道[30],NiO的衍射峰位于37.3°、43.3°、62.9°和75.4°,分别对应于其(111)、(200)、(220)和(311)晶面。从图 2(a)中没有看到明显的NiO的衍射峰,说明Ni物种在四种载体上高度分散。
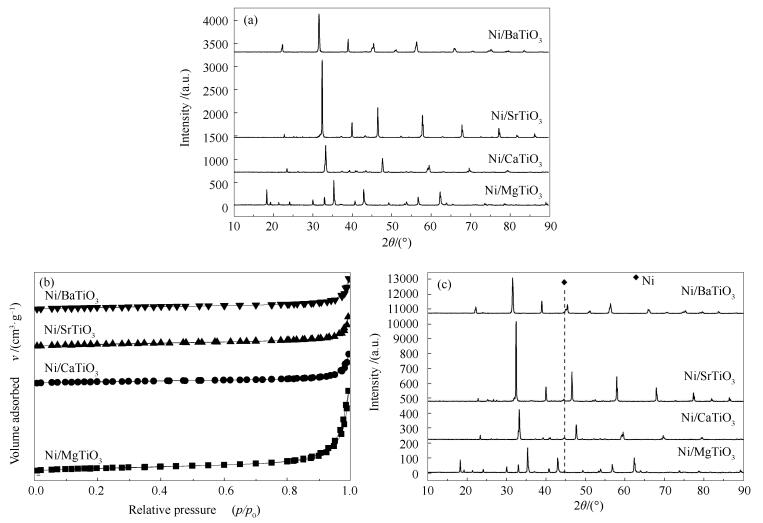
图 2(b)为制备的Ni催化剂的N2吸附等温线图。由图 2(b)和表 1可知,四种钙钛矿载体在引入活性组分后,其孔道结构并未受到明显影响,但对其比表面积和孔体积有着微弱影响。Ni/MgTiO3、Ni/SrTiO3和Ni/BaTiO3的比表面积和孔体积比相应的载体要大,说明NiO会在载体表面堆积形成一定的孔道结构;而Ni/CaTiO3的比表面积比CaTiO3小但是孔体积比CaTiO3大,表明有部分NiO进入了载体的孔道,有部分NiO在载体表面堆积。
图 2(c)为还原后Ni催化剂的XRD谱图。由图 2(c)可知,还原后催化剂与新鲜催化剂(图 2(a))归属于载体的XRD谱峰没有明显差异,说明还原过程没有对催化剂的晶型结构造成明显影响。然而,Ni/CaTiO3、Ni/BaTiO3、Ni/SrTiO3三种催化剂在44.5°处均出现了Ni的衍射峰,但是Ni/MgTiO3催化剂上没有明显的Ni衍射峰。
催化剂的活性和稳定性不仅与催化剂的物理性质(如:颗粒粒径等)有关,也与活性组分与载体的相互作用有关[27]。实验中采用H2-TPR研究了活性组分和载体间的相互作用。图 3为钙钛矿负载Ni催化剂的H2-TPR谱图。
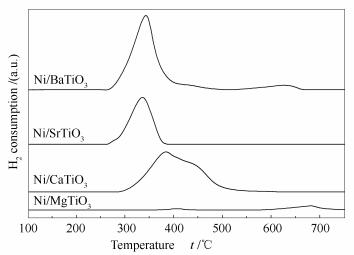
由图 3可知,四种催化剂的还原性差异较大,这说明它们表面的NiO物种的分散状态不同。对于Ni/MgTiO3催化剂,400℃左右的还原峰归属于与载体有着弱相互作用的NiO物种的还原。根据文献,在NiO/MgO催化剂中,500-750℃的还原峰为NiO-MgO固溶体的还原,由此判断出在600-700℃的还原峰归属于与载体形成NiO-MgO固溶体中Ni2+的还原[31]。对于Ni/CaTiO3催化剂,其H2-TPR谱图可以拆分为三个峰:低温、中温和高温还原峰,它们分别归属于载体表面孤立的NiO的还原、与载体有弱相互作用的NiO的还原、与载体有强相互作用的NiO的还原。对于Ni/SrTiO3和Ni/BaTiO3催化剂,320℃左右的还原峰可以归属于载体表面孤立的NiO的还原。而Ni/BaTiO3催化剂在600℃以上的还原峰也可以归属于与载体形成某种化合物的Ni物种的还原。从还原峰的温度来看,Ni/CaTiO3催化剂中金属载体相互作用更强,Ni/SrTiO3催化剂中的Ni物种与载体几乎没有相互作用;从还原峰的面积可以看出,四种催化剂的还原度也即还原出的活性Ni物种的数量,呈现出以下规律:Ni/CaTiO3>Ni/BaTiO3>Ni/SrTiO3>Ni/MgTiO3。
图 4是还原后Ni催化剂的CO2-TPD谱图。200℃以下、200-300℃和300℃以上的脱附峰可以分别归属为弱碱性位、中强碱性位和强碱性位上CO2的脱附。
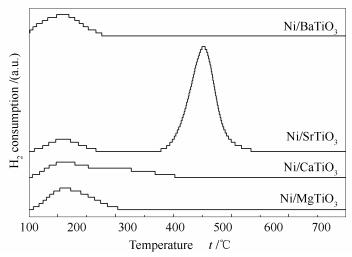
由图 4可知,Ni/MgTiO3和Ni/BaTiO3催化剂有着相似的脱附行为,脱附峰的温度在200℃以下,说明这两种催化剂只有弱碱性位;Ni/CaTiO3催化剂上有两个脱附峰,一个归属为弱碱性位,另一个归属为中强碱性位;Ni/SrTiO3催化剂上也有两个脱附峰,一个归属为弱碱性位,另一个归属为强碱性位。从碱性位的数量也即脱附峰面积大小来说,Ni/SrTiO3催化剂上碱性位的数量最多,其次是Ni/CaTiO3和Ni/MgTiO3,Ni/BaTiO3催化剂上碱性位的数量最少。
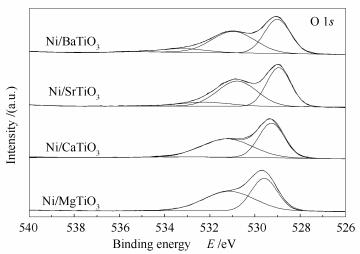
不同催化剂的O 1s的XPS谱图见图 5。由图 5可知,四种催化剂都具有两种类型的氧物种。结合能位于529eV附近的氧物种被认为是晶格氧物种(O2-),而结合能位于532eV附近的氧物种是吸附氧物种(O-/O2-/OH-)[32]。不同催化剂表面氧物种的分布存在较大差异。对谱图进行拟合后的结果见表 2。
 下载:
导出CSV
下载:
导出CSV
| Sample | Lattice oxygen | Ni elemental composition /% |
|
| EB/eV | area /% | ||
| Ni/MgTiO3 | 529.60 | 46.20 | 6.77 |
| Ni/CaTiO3 | 529.27 | 48.26 | 6.90 |
| Ni/SrTiO3 | 528.96 | 43.89 | 6.48 |
| Ni/BaTiO3 | 529.01 | 44.77 | 7.45 |
由表 2可知,不同催化剂的晶格氧物种的结合能不同,相比于Ni/MgTiO3催化剂,Ni/CaTiO3和Ni/BaTiO3催化剂晶格氧的结合能较低,说明Ni/CaTiO3和Ni/BaTiO3中晶格氧较为活泼,具有较高的迁移能力[33]。据文献报道[33-35],钙钛矿中的晶格氧物种在DRM反应中对甲烷分子的C-H键的活化起着关键的作用。晶格氧物种的结合能越低,则其与钙钛矿载体中其余组分的键合越弱,说明晶格氧物种更加活泼,迁移能力较强,便更易活化CH4分子中的C-H键。
图 6为四种催化剂的活性评价结果。为了更好地对四种催化剂的反应性能进行对比,反应温度设定在700℃。由图 6可知,四种催化剂的活性顺序为:Ni/CaTiO3 > Ni/BaTiO3 > Ni/MgTiO3 > Ni/SrTiO3。由出口气体的H2/CO体积比可以看出,四种催化剂表面的逆水煤气变换反应(RWGS)的剧烈程度不同。Ni/SrTiO3表面的RWGS反应最为强烈,Ni/MgTiO3次之,Ni/CaTiO3最弱。Ni是甲烷二氧化碳重整的活性组分,因此,催化剂在还原过程中暴露出的活性位的多少与催化剂的活性高低密切相关。图 3中H2-TPR的结果表明,Ni/CaTiO3和Ni/BaTiO3催化剂还原出的Ni物种最多,表 2中XPS表面原子相对含量分析结果表明,Ni/CaTiO3和Ni/BaTiO3催化剂表面活性组分Ni的相对原子含量较多。这可能是这两个催化剂活性较高的主要原因。此外,Ni/CaTiO3和Ni/BaTiO3催化剂更为活泼的晶格氧也是其初始活性较高的关键因素。由于Ni/MgTiO3催化剂形成了NiO-MgO固溶体,使得活性组分较难还原,催化剂表面可利用的活性组分Ni物种的相对含量较低,并且晶格氧物种不够活泼,流动性较差,对CH4分子中C-H键的活化能力较低,使得Ni/MgTiO3催化剂上CH4和CO2的转化率比前两者低。对于Ni/SrTiO3催化剂,由XRD谱图可知,SrTiO3载体颗粒粒径较大,很可能造成其Ni的分散不高;且活性组分和载体的相互作用较弱(H2-TPR),Ni的表面原子相对含量较低(XPS),晶格氧活化C-H键的能力较弱(XPS)。这应该是造成其低活性的原因。Ni/MgTiO3催化剂活性高于Ni/SrTiO3催化剂可能与它们表面Ni的分散度有关。同时也可以看到,Ni/CaTiO3和Ni/MgTiO3催化剂的失活要比Ni/BaTiO3慢。
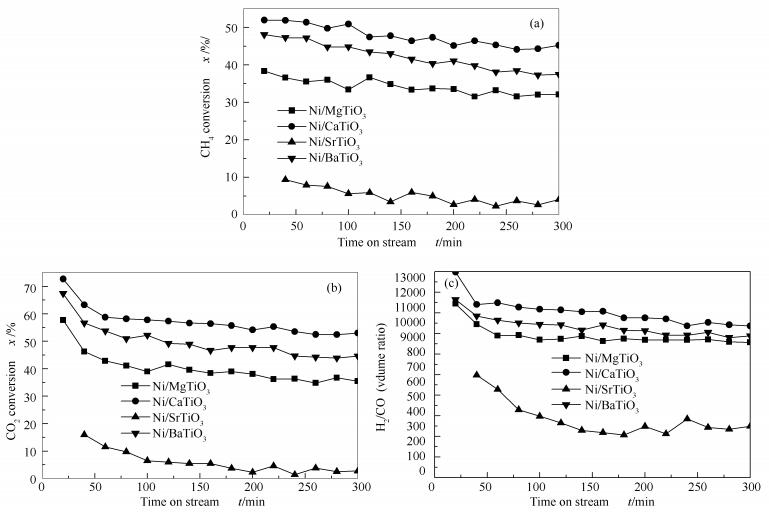
图 7(a)为反应后催化剂的XRD谱图。与图 2(c)相比,Ni/BaTO3催化剂于26.2°处出了现明显的碳的衍射峰,表明反应后的催化剂上有晶相石墨炭的存在[36]。说明Ni/BaTiO3催化剂较其余三种催化剂,在反应过程中生成了较多的且活性低的积炭。
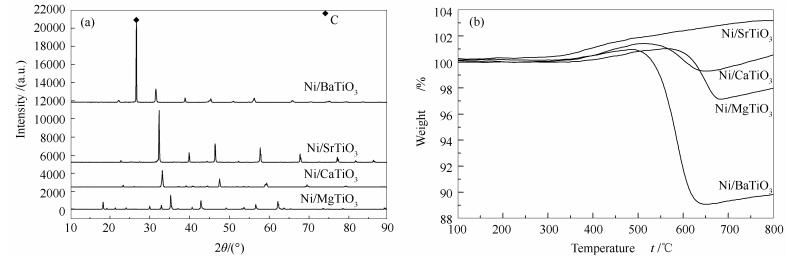
图 7(b)为反应后催化剂的TG曲线。由图 7(b)可知,Ni/BaTiO3催化剂上积炭最多,这与图 7(a)得出的结论一致,这可能是Ni/BaTiO3催化剂失活最快的原因。Ni/SrTiO3催化剂上几乎没有发生积炭,这是因为该催化剂上CH4几乎没有转化。Ni/CaTiO3催化剂上的积炭比Ni/MgTiO3催化剂少。由前文CO2-TPD的结果可知,Ni/CaTiO3催化剂具有更多的碱性位,因此,其对CO2的吸附活化能力更强,也即该催化剂的消炭能力较强,导致其表面有较少积炭。
通过改变A位元素调变ABO3型钙钛矿复合氧化物的性质,用浸渍法制备了一系列负载型Ni/MTiO3 (M=Mg、Ca、Sr、Ba)催化剂用于DRM反应,考察了载体性质的改变对催化剂上DRM反应性能的影响。结果表明,A位元素为不同碱土金属时,催化剂活性组分和载体的相互作用,活性组分的表面原子浓度以及催化剂晶格氧的流动性都发生了改变。Ni/CaTiO3催化剂由于载体和活性组分的相互作用较强,可还原出的活性组分Ni的含量较多,且晶格氧流动性较高有较好的活化CH4分子中C-H键的能力,因而在四种催化剂中表现出最高的催化性能;Ni/BaTiO3由于碱性较Ni/CaTiO3弱,活化CO2的能力较差,积炭较为严重,催化剂失活较快;Ni/MgTiO3上形成了NiO-MgO固溶体,活性组分难以被还原,DRM反应活性较差;SrTiO3载体颗粒粒径较大,Ni/SrTiO3催化剂上Ni的分散度不高,活性组分和载体的相互作用较弱,且Ni的表面原子相对含量较低,晶格氧的流动性较差导致了Ni/SrTiO3催化剂上最低的DRM活性。
NEJAT P, JOMEHZADEH F, TAHERI M M, GOHARI M, ABD MAJID M Z. A global review of energy consumption, CO2 emissions and policy in the residential sector (with an overview of the top ten CO2 emitting countries)[J]. Renewable Sustainable Energy Rev, 2015, 43: 843-862.
王辉, 张俊峰, 白云星, 王文峰, 谭猗生, 韩怡卓. NiO@SiO2核壳催化剂在浆态床中低温甲烷化研究[J]. 燃料化学学报, 2016,44,(5): 548-556. WANG Hui, ZHANG Jun-feng, BAI Yun-xing, WANG Wen-feng, TAN Yi-sheng, HAN Yi-zhuo. NiO@SiO2 core-shell catalyst for low-temperature methanation of syngas in slurry reactor[J]. J Fuel Chem Technol, 2016, 44(5): 548-556.
石广强, 李君华, 刘宇航, 武则龙. 甲烷间接转化制合成气的研究进展[J]. 天津化工, 2015,29,(4): 1-3. SHI Guang-qiang, LI Jun-hua, LIU Yu-hang, WU Ze-long. Research progress on indirect conversion of methane to produce synthesis gas[J]. Tianjin Chem Ind, 2015, 29(4): 1-3.
张小平. 介孔Ni/ZrO2催化剂的制备及甲烷二氧化碳重整催化性能[J]. 山东化工, 2019,48,(10): 43-45. ZHANG Xiao-ping. Preparation of mesoporous Ni/ZrO2 catalysts and catalytic performance for CO2 reforming of CH4[J]. Shandong Chem Ind, 2019, 48(10): 43-45.
HERMAN R G, SUN Q, SHI C. Development of active oxide catalysts for the direct oxidation of methane to formaldehyde[J]. Catal Today, 1997, 37(1): 1-14.
XU J, ZHANG Y, XU X, FANG X, XI R, LIU Y, ZHENG R, WANG X. Constructing La2B2O7 (B=Ti, Zr, Ce) compounds with three typical crystalline phases for oxidative coupling of methane:The effect of phase structures, super oxide anions and alkalinity on the reactivity[J]. ACS Catal, 2019, 9(5): 4030-4045.
GUO X, FANG G, LI G, MA H, FAN H, YU L, MA C, WU X, DENG D, WEI M. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen[J]. Science, 2014, 344(2): 616-619.
ROSTRUP-NIELSEN J R. Sulfur-passivated nickel catalysts for carbon-free steam reforming of methane[J]. J Catal, 1984, 85(1): 31-43.
XIAO Z, LI Y, HOU F, WU C, PAN L, ZOU J, WANG L, ZHANG X, LIU G, LI G. Engineering oxygen vacancies and nickel dispersion on CeO2 by Pr doping for highly stable ethanol steam reforming[J]. Appl Catal B:Environ, 2019, 258: 117940.
YANG J H, YOON Y, RYU M, AN S K, SHIN J, LEE C J. Integrated hydrogen liquefaction process with steam methane reforming by using liquefied natural gas cooling system[J]. Appl Energy, 2019, 255: 113840.
DI GIULIANO A, GALLUCCI K, GIANCATERINO F, COURSON C, FOSCOLO P U. Multicycle sorption enhanced steam methane reforming with different sorbent regeneration conditions:Experimental and modelling study[J]. Chem Eng J, 2019, 377: 119874.
MA Y, MA Y, LONG G, LI J, HU X, YE Z, WANG Z, BUCKLEY C E, DONG D, Synergistic promotion effect of MgO and CeO2 on nanofibrous Ni/Al2O3 catalysts for methane partial oxidation[J]. Fuel, 2019, 258: 116103. https://www.researchgate.net/publication/337662135_Synergistic_promotion_effect_of_MgO_and_CeO2_on_nanofibrous_NiAl2O3_catalysts_for_methane_partial_oxidation
KIM S, CRANDALL B S, LANCE M J, CORDONNIER N, LAUTERBACH J, SASMAZ E. Activity and stability of NiCe@SiO2 multi-yolk-shell nanotube catalyst for tri-reforming of methane[J]. Appl Catal B:Environ, 2019, 259: 118037.
ZHANG X, ZHANG M, ZHANG J, ZHANG Q, TSUBAKI N, TAN Y, HAN Y. Methane decomposition and carbon deposition over Ni/ZrO2 catalysts:Comparison of amorphous, tetragonal, and monoclinic zirconia phase[J]. Int J Hydrogen Energy, 2019, 44(33): 17887-17899.
ZHAO Y, KANG Y, LI H, LI H. CO2 conversion to synthesis gas via DRM on the durable Al2O3/Ni/Al2O3 sandwich catalyst with high activity and stability[J]. Green Chem, 2018, 20(12): 2781-2787.
QIN Z, CHEN J, XIE X, LUO X, SU T, JI H. CO2 reforming of CH4 to syngas over nickel-based catalysts[J]. Environ Chem Lett, 2020, 18(4): 997-1017.
ZHANG M, ZHANG J, WU Y, PAN J, ZHANG Q, TAN Y, HAN Y. Insight into the effects of the oxygen species over Ni/ZrO2 catalyst surface on methane reforming with carbon dioxide[J]. Appl Catal B:Environ, 2019, 244: 427-437.
YENTEKAKIS I V, GOULA G, HATZISYMEON M, BETSI-ARGYROPOULOU I, BOTZOLAKI G, KOUSI K, KONDARIDES D I, TAYLOR M J, PARLETT C. M. A, OSATIASHTIANI A, KYRIAKOU G, HOLGADO J P, LAMBERT R M. Effect of support oxygen storage capacity on the catalytic performance of Rh nanoparticles for CO2 reforming of methane[J]. Appl Catal B:Environ, 2019, 243: 490-501.
LIU C, YE J, JIANG J, PAN Y. Progresses in the preparation of coke resistant Ni-based catalyst for steam and CO2 reforming of methane[J]. Chem Cat Chem, 2011, 3(3): 529-541.
ARBAG H, YASYERLI S, YASYERLI N, DOGU G. Activity and stability enhancement of Ni-MCM-41 catalysts by Rh incorporation for hydrogen from dry reforming of methane[J]. Int J Hydrogen Energy, 2010, 35(6): 2296-2304.
HU Y H, RUCKENSTEIN E. Binary MgO-based solid solution catalysts for methane conversion to syngas[J]. Catal Rev, 2002, 44(3): 423-453.
AZIZ M.A.A, JALIL A. A, WONGSAKULPHASATCH S, VO D V N. Understanding the role of surface basic sites of catalysts in CO2 activation in dry reforming of methane:A short review[J]. Catal Sci Technol, 2020, 10(1): 35-45.
WANG S, LU G Q, MILLAR G J. Carbon Dioxide reforming of methane to produce synthesis gas over metal-supported catalysts:State of the art[J]. Energy Fuels, 1996, 10(4): 896-904.
LI Z, WANG Z, JIANG B, KAWI S. Sintering resistant Ni nanoparticles exclusively confined within SiO2 nanotubes for CH4 dry reforming[J]. Catal Sci Technol, 2018, 8(13): 3363-3371.
ZHANG M, ZHANG J, ZHOU Z, CHEN S, ZHANG T, SONG F, ZHANG Q, TSUBAKI N, TAN Y, HAN Y. Effects of the surface adsorbed oxygen species tuned by rare-earth metal doping on dry reforming of methane over Ni/ZrO2 catalyst[J]. Appl Catal B:Environ, 2020, 264: 118522.
YAN Q G, WENG W Z, WAN H L, TOGHIANI H, JR C U P. Activation of methane to syngas over a Ni/TiO2 catalyst[J]. Appl Catal A:Gen, 2003, 239(1/2): 43-58.
AKRI M, ZHAO S, LI X, ZANG K, LEE A F, ISAACS M A, XI W, GANGARAJULA Y, LUO J, REN Y, CUI Y T, LI L, SU Y, PAN X, WEN W, PAN Y, WILSON K, LI L, QIAO B, ISHII H, LIAO Y F, WANG A, WANG X, ZHANG T. Atomically dispersed nickel as coke-resistant active sites for methane dry reforming[J]. Nat Commun, 2019, 10(1): 5181.
HAYAKAWA T, SUZUKI S, NAKAMURA J, UCHIJIMA T, HAMAKAWA S, SUZUKI K, SHISHIDO T, TAKEHIRA K. CO2 reforming of CH4 over Ni/perovskite catalysts prepared by solid phase crystallization method[J]. Appl Catal A:Gen, 1999, 183(2): 273-285.
LI X, WU M, LAI Z, HE F. Studies on nickel-based catalysts for carbon dioxide reforming of methane[J]. Appl Catal A:Gen, 2005, 290(1/2): 81-86.
GAO N, CHENG M, QUAN C, ZHENG Y. Syngas production via combined dry and steam reforming of methane over Ni-Ce/ZSM-5 catalyst[J]. Fuel, 2020, 273: 117702.
WANG Y H, LIU H M, XU B Q. Durable Ni/MgO catalysts for CO2 reforming of methane:Activity and metal-support interaction[J]. J Mol Catal A:Chem, 2009, 299(1): 44-52.
LÖFBERG A, GUERRERO-CABALLERO J, KANE T, RUBBENS A, JALOWIECKI-DUHAMEL L. Ni/CeO2 based catalysts as oxygen vectors for the chemical looping dry reforming of methane for syngas production[J]. Appl Catal B:Environ, 2017, 212: 159-174.
SUTTHIUMPORN K, MANEERUNG T, KATHIRASER Y, KAWI S. CO2 dry-reforming of methane over La0.8Sr0.2Ni0.8M0.2O3 perovskite (M=Bi, Co, Cr, Cu, Fe):Roles of lattice oxygen on C-H activation and carbon suppression[J]. Int J Hydrogen Energy, 2012, 37(15): 11195-11207.
LI R J, YU C C, JI W J, SHEN S K. Methane oxidation to synthesis gas using lattice oxygen in La1-xSrxFeO3 perovskite oxides instead of molecular oxygen[J]. Stud Surf Sci Catal, 2004, 147: 199-204.
AU C T, HU Y H, WAN H L. Methane activation over unsupported and La2O3-supported copper and nickel catalysts[J]. Catal Lett, 1996, 36(3/4): 159-163.
LIU B S, AU C T. Carbon deposition and catalyst stability over La2NiO4/γ-Al2O3 during CO2 reforming of methane to syngas[J]. Appl Catal A Gen, 2003, 244(1): 181-195.

图 1 钙钛矿载体的XRD谱图(a)和N2吸附等温线(b)
Figure 1 XRD patterns (a) and N2 sorption isotherms (b) of various MTiO3 perovskite supports

图 2 新鲜催化剂的XRD谱图(a)和N2吸附等温线(b), 还原后催化剂的XRD谱图(c)
Figure 2 XRD patterns (a) and N2 sorption isotherms (b) of the fresh Ni/MTiO3 catalysts as well as the XRD patterns of the reduced Ni/MTiO3 catalysts (c)

图 3 Ni/MTiO3(M=Mg、Ca、Sr、Ba)催化剂的H2-TPR谱图
Figure 3 H2-TPR profiles of Ni/MTiO3 (M=Mg, Ca, Sr, Ba) catalysts

图 4 Ni/MTiO3(M=Mg、Ca、Sr、Ba)催化剂的CO2-TPD谱图
Figure 4 CO2-TPD profiles of the Ni/MTiO3 catalysts (M=Mg, Ca, Sr, Ba)

图 5 还原后的Ni/MTiO3(M=Mg、Ca、Sr、Ba)催化剂的XPS谱图
Figure 5 XPS spectra of the reduced Ni/MTiO3(M=Mg, Ca, Sr, Ba) catalysts

图 6 Ni/MTiO3(M=Mg、Ca、Sr、Ba)催化剂的DRM反应性能
Figure 6 Performance of the Ni/MTiO3 catalysts (M = Mg, Ca, Sr, Ba) in DRM

图 7 反应后Ni/MTiO3(M=Mg、Ca、Sr、Ba)催化剂的XRD谱图(a)和热重曲线(b)
Figure 7 XRD patterns (a) and TG profiles (b) of the spent Ni/MTiO3 (M=Mg, Ca, Sr, Ba) catalysts
表 1 钙钛矿载体及负载催化剂的织构性质
Table 1. Textural properties of the perovskite supports and supported Ni catalysts
| Sample | Surface area A/(m2·g-1) |
Pore volume v/(cm3·g-1) |
Crystalline sizea /nm |
Sample | Surface area A/(m2·g-1) |
Pore volume v/(cm3·g-1) |
| MgTiO3 | 4.98 | 0.0106 | 44.0 | Ni/MgTiO3 | 6.05 | 0.0190 |
| CaTiO3 | 3.65 | 0.0049 | 34.0 | Ni/CaTiO3 | 3.05 | 0.0065 |
| SrTiO3 | 3.53 | 0.0048 | 53.2 | Ni/SrTiO3 | 4.04 | 0.0070 |
| BaTiO3 | 2.56 | 0.0039 | 34.6 | Ni/BaTiO3 | 3.20 | 0.0067 |
| a: calculated by Scherrer equation according to the XRD results | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 还原后的Ni/MTiO3(M=Mg、Ca、Sr、Ba)的催化剂的XPS拟合
Table 2. XPS fitting results of the reduced Ni/MTiO3 (M=Mg, Ca, Sr, Ba) catalysts
| Sample | Lattice oxygen | Ni elemental composition /% |
|
| EB/eV | area /% | ||
| Ni/MgTiO3 | 529.60 | 46.20 | 6.77 |
| Ni/CaTiO3 | 529.27 | 48.26 | 6.90 |
| Ni/SrTiO3 | 528.96 | 43.89 | 6.48 |
| Ni/BaTiO3 | 529.01 | 44.77 | 7.45 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们