引用本文:
邱泽刚, 李侨, 马少博, 李志勤. 碳化终温对β-Mo2C催化喹啉加氢脱氮性能影响[J]. 燃料化学学报,
2020, 48(3): 357-368.
Citation:
QIU Ze-gang, LI Qiao, MA Shao-bo, LI Zhi-qin. Effect of final carbonization temperature on catalytic performance of β-Mo2C in quinoline hydrodenitrogenation[J]. Journal of Fuel Chemistry and Technology,
2020, 48(3): 357-368.
Received Date:
20 December 2019 Revised Date:
30 January 2020 Available Online:
01 March 2020
Fund Project:
The project was supported by the National Natural Science Foundation of China (21878243, 21606177) and Shanxi Natural Science Basic Research Program (2019JM-085)
Abstract:
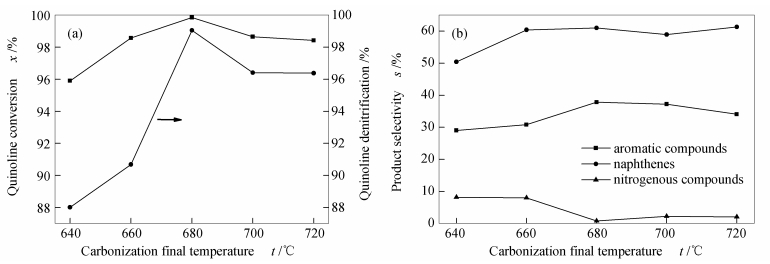
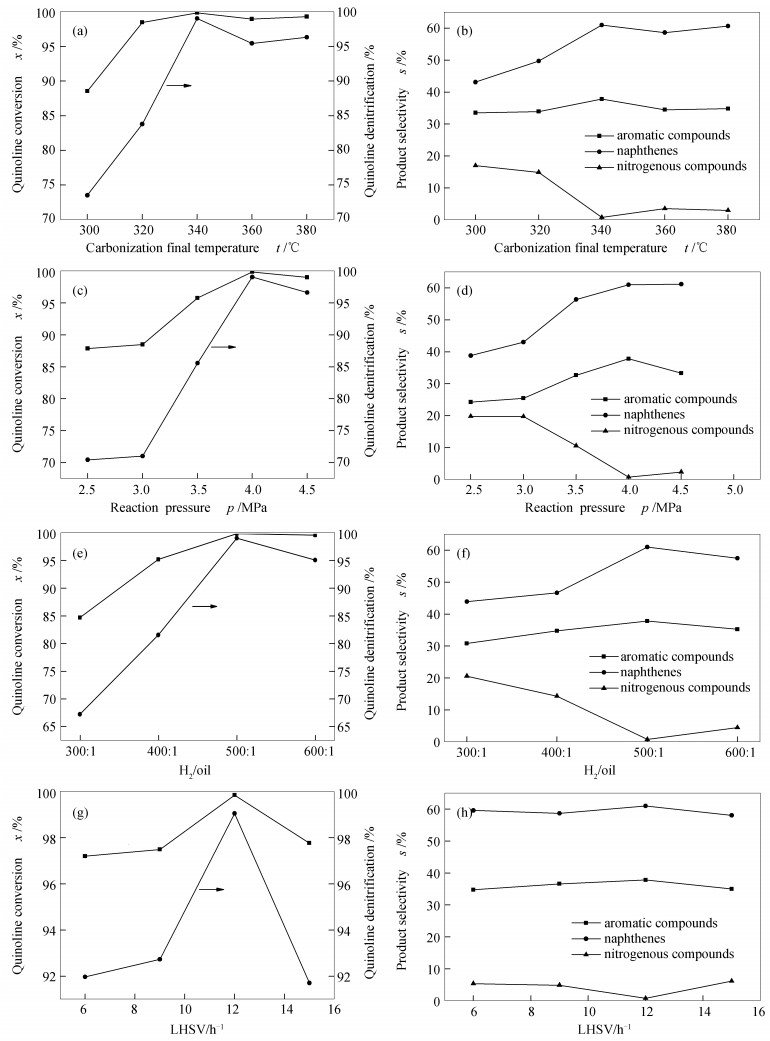
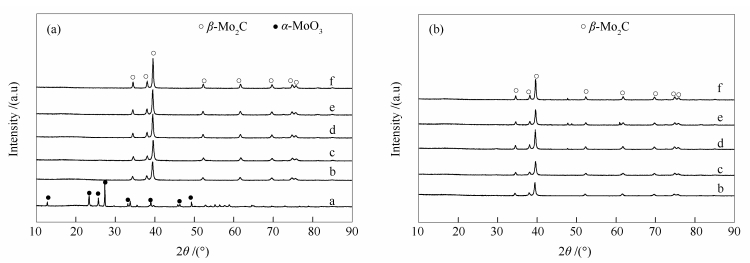
MoO3 was used as precursor, CH4/H2 as carbon source, and a direct reduction carbonization method with programmed temperature rise was used to prepare molybdenum carbide catalysts at different final carbonization temperatures (640, 660, 680, 700, and 720℃). The physical properties and structural properties of molybdenum carbide were characterized by XRD, N2 adsorption, SEM, TEM, XPS and Raman. The effect of final carbonization temperature on the catalytic performance of molybdenum carbide in quinoline hydrodenitrogenation was studied. The results showed that the molybdenum carbide catalysts with different final carbonization temperatures were all existed in the phase of β-Mo2C. The final carbonization temperature could significantly change content of species on the surface, average pore size, and mesopore distribution of molybdenum carbide. When the final carbonization temperature was 680℃, a higher carbonization degree, the lowest content of oxygen species on the surface and the highest surface C/Mo molar ratio of catalyst were obtained; accordingly, the best catalytic activity of catalysts was achieved. At 340℃ and 4 MPa, the conversion and denitrification rate of quinoline were up to 99%, while the selectivity of aromatic compounds was up to 37.8%, showing a lower aromatic ring destruction. Surface composition, especially surface oxygen, was essential for the regulation of the quinoline hydrodenitrogenation reaction pathway on β-Mo2C.
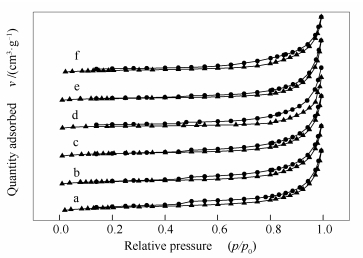
Figure 2.
Effect of different final carbonization temperatures on the performance of molybdenum carbide catalystsreaction conditions: t=340 ℃, p=4.0 MPa, LHSV=12 h-1, H2/oil=500
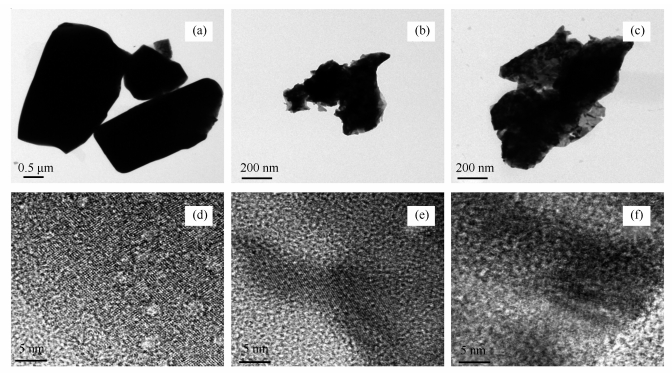
(a), (d): TEM and HRTEM photos of MoO3; (b), (e): TEM and HRTEM photos of 680 ℃ (before reaction); (c), (f): TEM and HRTEM photos of 680 ℃ (after reaction)
Figure 7.
TEM and HRTEM photos of MoO3 and molybdenum carbide catalyst before and after reaction
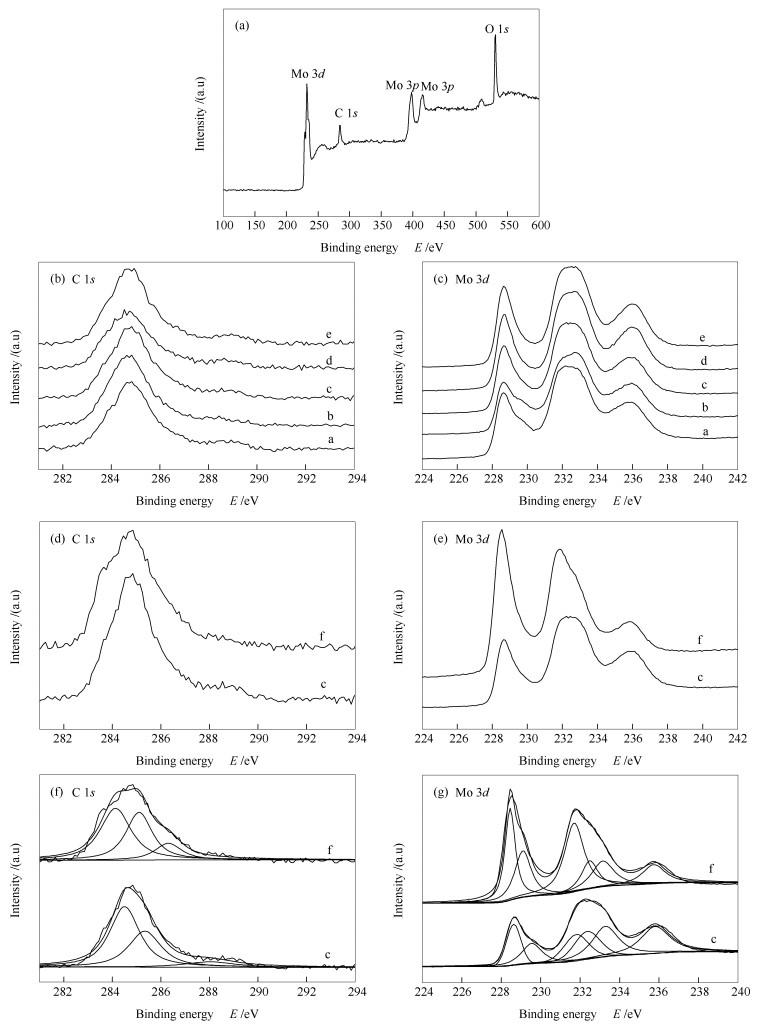
(a): wide scan XPS spectrum of molybdenum carbide; (b), (c): XPS spectra of molybdenum carbide catalysts with different final carbonization temperatures; (d), (e): high-resolution XPS spectra of molybdenum carbide before and after the reaction; (f), (g): high resolution XPS decomposition spectra of molybdenum carbide before and after the reaction a: 640 ℃; b: 660 ℃; c: 680 ℃; d: 700 ℃; e: 720 ℃; f: 680 ℃(after reaction)
Figure 8.
XPS spectra of molybdenum carbide catalyst
马宝歧, 任沛建, 杨占彪.煤焦油制燃料油品[M].北京:化学工业出版社, 2011.MA Bao-qi, REN Pei-jian, YANG Zhan-biao. Fuel Oil from Coal Tar Oil[M]. Beijing:Chemical Industry Press, 2011.
[2]
李大东.加氢处理工艺与工程[M].北京:中国石化出版社, 2004, 170-435.LI Da-dong. Hydrotreating Process and Engineering[M]. Beijing:China Petrochemical Press, 2004, 170-435.
[3]
石垒, 张增辉, 邱泽刚, 郭芳, 张伟, 赵亮富. P改性对Mo-Ni/Al2O3煤焦油加氢脱氧性能的影响[J]. 燃料化学学报,
2015,43,(1): 74-80.
doi: 10.3969/j.issn.0253-2409.2015.01.012SHI Lei, ZHANG Zeng-hui, QIU Ze-gang, GUO Fang, ZHANG Wei, ZHAO Liang-fu. Effect of phosphorus modification on the catalytic properties of Mo-Ni/Al2O3 in the hydrodenitrogenation of coal tar[J]. J Fuel Chem Technol,
2015, 43(1):
74-80.
doi: 10.3969/j.issn.0253-2409.2015.01.012
[4]
胡乃方, 崔海涛, 邱泽刚, 赵亮富, 孟欣欣, 赵正权, 敖广宇. 不同P负载量对Co-Mo/γ-Al2O3煤焦油加氢脱硫性能影响的研究[J]. 燃料化学学报,
2016,44,(6): 745-753.
doi: 10.3969/j.issn.0253-2409.2016.06.016HU Nai-fang, CUI Hai-tao, QIU Ze-gang, ZHAO liang-fu, MENG Xin-xin, ZHAO Zheng-quan, AO Guang-yu. Effect of phosphorus loadings on the performance of Co-Mo/γ-Al2O3 in hydrodesulfurization of coal tar[J]. J Fuel Chem Technol,
2016, 44(6):
745-753.
doi: 10.3969/j.issn.0253-2409.2016.06.016
[5]
NIU M, SUN X, LI D, CUI W G, ZHANG X, BAI X X, LI W H. The hydrodeoxygenation, hydrogenation, hydrodealkylation and ring-opening reaction in the hydrotreating of low temperature coal tar over Ni-Mo/γ-Al2O3 catalyst[J]. React Kinet Mech Catal,
2017, 121(6):
1-17.
[6]
TANG W, FANG M, WANG H, YU P, WANG Q, LUO Z. Mild hydrotreatment of low temperature coal tar distillate, Product composition[J]. Chem Eng J,
2014, 236(2):
529-537.
XIA L Y, XIA Z X, TANG W, WANG H Y, FANG M X. Hydrogenation of model compounds catalyzed by MCM-41-supported nickel phosphide[J]. Adv Mater Res,
2014, 864:
366-372.
[9]
HAN W, NIE H, LONG X Y, LI M F, YANG Q H, LI D D. Preparation of F-doped MoS2/Al2O3 catalysts as a way to understand the electronic effects of the support Brønsted acidity on HDN activity[J]. J Catal,
2016, 339:
135-142.
doi: 10.1016/j.jcat.2016.04.005
[10]
SHAO M Q, CUI H T, GUO S Q, ZHAO L F, TAN Y S. Effects of calcination and reduction temperature on the properties of Ni-P/SiO2 and Ni-P/Al2O3 and their hydrodenitrogenation performance[J]. Rsc Adv,
2018, 8(13):
6745-6751.
doi: 10.1039/C7RA11907K
[11]
NGUYEN M T, TAYAKOUTFAYOLLE M, PIRNGRUBER G D, CHAINET F, GEANTET C. Kinetic modeling of quinoline hydrodenitrogenation over a NiMo(P)/Al2O3 catalyst in a batch reactor[J]. Ind Eng Chem Res,
2015, 54(38):
9278-9288.
doi: 10.1021/acs.iecr.5b02175
[12]
WANG W, LI X, SUN Z C, WANG A J, LIU Y Y, CHEN Y Y, D C P. Influences of calcination and reduction methods on the preparation of Ni2P/SiO2 and its hydrodenitrogenation performance[J]. Appl Catal A:Gen,
2016, 509(1):
45-51.
[13]
SCHLATTER J C, OYAMA S T, METCALFE J E. Catalytic behavior of selected transition metal carbides, nitrides, and borides in the hydrodenitrogenation of quinoline[J]. Ind Eng Chem Res,
1988, 27(9):
1648-1653.
doi: 10.1021/ie00081a014
[14]
DOLCE G M, THOMPSON L T. Supported molybdenum carbide catalysts, structure-function relationships for hydrodenitrogenation[J]. Mat Res Soc Symp Proc,
1996, 454:
47-52.
doi: 10.1557/PROC-454-47
[15]
CHI J Q, GAO W K, LIN J H, DONG B, QIN J F, LIU Z Z, LIU B, CHAI Y M, LIU C G. Porous core-shell N-doped Mo2C@C nanospheres derived from inorganic-organic hybrid precursors for highly efficient hydrogen evolution[J]. J Catal,
2018, 360:
9-19.
doi: 10.1016/j.jcat.2018.01.023
[16]
CHI J Q, LIN J H, QIN J F, DONG B, YAN K L, LIU Z Z, ZHANG X Y, CHAI Y M, LIU C G. A triple synergistic effect from pitaya-like MoNix-MoCx hybrids ncapsulated in N-doped C nanospheres for efficient hydrogen evolution. Sustain[J]. Sustainable Energy Fuels,
2018, 2:
1610-1620.
doi: 10.1039/C8SE00135A
[17]
JIAN M, PRINS R. Mechanism of the hydrodenitrogenation of quinoline over NiMo(P)/Al2O3 catalysts[J]. J Catal,
1998, 113(1):
111-123.
[18]
SEBAKHY K O, VITALE G, HASSAN A, PEREIRA-ALMAO P. New insights into the kinetics of structural transformation and hydrogenation activity of nano-crystalline molybdenum carbide[J]. Catal Lett,
2018, 148(3):
904-923.
doi: 10.1007/s10562-017-2274-3
[19]
GUO F, GUO S Q, WEI X X, WANG X X, XIANG H W, QIU Z G, ZHAO L F. The effects of MCM-41's calcination temperature on the structure and hydrodenitrogenation over NiW catalysts[J]. Korean J Chem Eng,
2014, 31(11):
1973-1979.
doi: 10.1007/s11814-014-0148-6
[20]
黄澎. SBA-15负载磷化镍催化剂对喹啉加氢脱氮反应网络的影响[J]. 煤炭学报,
2015,40,(2): 195-200.
HUANG Peng. Effect of SBA-15-supported nickle phosphate catalyst on hydrodenitrogenation network of quinoline[J]. J China Coal Soc,
2015, 40(2):
195-200.
[21]
尹海亮, 刘新亮, 周同娜, 赵健, 蔺爱国. 乙二醇对磷掺杂NiMo/Al2O3加氢催化剂性能的影响[J]. 燃料化学学报,
2019,47,(12): 1459-1467.
YIN Hai-liang, LIU Xin-liang, ZHOU Tong-na, ZHAO Jian, LIN Ai-guo. Effect of ethylene glycol on the hydrogenation performance of P-doped NiMo/Al2O3 catalysts[J]. J Fuel Chem Technol,
2019, 47(12):
1459-1467.
[22]
向明林, 李德宝, 肖海成, 张建利, 李文怀, 钟炳, 孙予罕. 碳化钼催化材料的制备、表征及CO加氢反应性能的研究[J]. 燃料化学学报,
2007,35,(3): 324-328.
doi: 10.3969/j.issn.0253-2409.2007.03.014XIANG Ming-lin, LI De-bao, XIAO Hai-cheng, ZHANG Jian-li, LI Wen-huai, ZHONG Bing, SUN Yu-han. Preparation and characterization of molybdenum carbide and its performance on the hydrogenation of carbon monoxide[J]. J Fuel Chem Technol,
2007, 35(3):
324-328.
doi: 10.3969/j.issn.0253-2409.2007.03.014
[23]
LI S, CHENG C, SAGALTCHIK A, PACHFULE P, ZHAO C S, THOMAS A. Metal-organic precursor-derived mesoporous carbon spheres with homogeneously distributed molybdenum carbide/nitride nanoparticles for efficient hydrogen evolution in alkaline media[J]. Adv Funct Mater,
2019, 29:
18074199.
[24]
ZHOU G Y, YANG Q, GUO X M, CHEN Y, YANG Q, XU L, SUN D M, TANG Y W. Coupling molybdenum carbide nanoparticles with N-doped carbon nanosheets as a high-efficiency electrocatalyst for hydrogen evolution reaction[J]. Int J Hydrogen Energy,
2018, 43:
9326-9333.
doi: 10.1016/j.ijhydene.2018.04.002
[25]
WAN J, WU J B, GAO X, TIAN Q L, HU Z M, YU H M, HUANG L. Structure confined porous Mo2C for efficient hydrogen evolution[J]. Adv Funct Mater,
2017, 27:
1703933.
doi: 10.1002/adfm.201703933
[26]
MAO T, XU J, YANG Y, LI Y W. Effect of carburization protocols on molybdenum carbide synthesis and study on its performance in CO hydrogenation[J]. Catal Today,
2016, 261:
101-115.
doi: 10.1016/j.cattod.2015.07.014
[27]
LEE W S, KUMAR A, WANG Z S, WANG X X, BHAN A. Chemical titration and transient kinetic studies of site requirements in Mo2C-catalyzed vapor phase anisole aydrodeoxygenation[J]. ACS Catal,
2015, 5:
4104-4114.
doi: 10.1021/acscatal.5b00713
[28]
SCHAIDLE J A, BLACKBURN J, FARBEROW C A, NASH C, STEIRER K X, CLARK J, RUDDY D A, ROBICHAUD D J. Experimental and computational investigation of acetic acid deoxygenation over oxophilic molybdenum carbide:Surface chemistry and active site identity[J]. ACS Catal,
2016, 6:
1181-1197.
doi: 10.1021/acscatal.5b01930
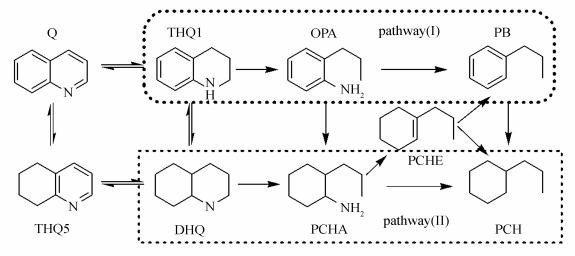
图 1
喹啉的HDN反应网络及反应途径示意图
Figure 1
HDN reaction network and reaction pathway of quinoline
Figure 2
Effect of different final carbonization temperatures on the performance of molybdenum carbide catalystsreaction conditions: t=340 ℃, p=4.0 MPa, LHSV=12 h-1, H2/oil=500
Figure 7
TEM and HRTEM photos of MoO3 and molybdenum carbide catalyst before and after reaction
(a), (d): TEM and HRTEM photos of MoO3; (b), (e): TEM and HRTEM photos of 680 ℃ (before reaction); (c), (f): TEM and HRTEM photos of 680 ℃ (after reaction)
Figure 8
XPS spectra of molybdenum carbide catalyst
(a): wide scan XPS spectrum of molybdenum carbide; (b), (c): XPS spectra of molybdenum carbide catalysts with different final carbonization temperatures; (d), (e): high-resolution XPS spectra of molybdenum carbide before and after the reaction; (f), (g): high resolution XPS decomposition spectra of molybdenum carbide before and after the reaction a: 640 ℃; b: 660 ℃; c: 680 ℃; d: 700 ℃; e: 720 ℃; f: 680 ℃(after reaction)

 下载:
下载:


 下载:
下载:










