
图 1.
LaCo1-xGaxO3样品(x=0、0.2、0.3、0.4和0.5)的XRD谱图
Figure 1.
XRD patterns of the LaCo1-xGaxO3 samples (x=0, 0.2, 0.3, 0.4 and 0.5)

鉴于化石燃料紧缺和温室效应问题的紧迫性,将丰富又廉价的CO2转化成高附加值的化学品的课题研究,引起全球研究者的广泛关注。但因其C=O键的断裂需要克服高能垒,CO2难以被活化[1],所以, 目前CO2的转化研究仍处于初步阶段。
近年来,CO2资源化利用的主要方向是通过加氢生成有附加值的化学品,包括CO2加氢生成CH4[2-4]、长链烃[2, 5]、甲醇(MeOH)[2, 5]、二甲醚(DME)[6]以及乙醇(EtOH)[7, 8]和低碳醇(HAs)[9]等。其中,加氢生成醇类(目前研究成果主要为合成MeOH方向)的反应是CO2转化利用最有前景的途径之一[1]。比起MeOH,EtOH具有高热值、低毒、运输安全以及应用广泛等优点[1, 2, 10]。但是相比于合成MeOH,需要催化剂具备双活性位点来完成C-C链增长和CO2部分还原[11, 12]步骤的EtOH合成过程更具挑战性。
目前, 所报道的用于CO2加氢制EtOH或HAs的催化剂主要有贵金属(Ru基[9]、Pt基[11]),Co基[13],Fe基[14]以及Cu基[15]催化剂体系,其中, 贵金属催化剂因其高催化活性和高HAs选择性而备受关注,如均相催化剂[Ru3(CO)12/Rh2(CO)4Cl2-LiI][9]在200 ℃下EtOH和HAs总选择性高达96.4%,但催化剂高昂的价格限制其在工业生产中的应用。而用非均相催化剂催化反应时,反应所需的高温促进了副反应逆水煤气反应(RWGS)的进行,使副产物CO增多。如Guo等[15]报道的K/Cu-Zn催化剂在350 ℃、3 MPa的反应条件下的CO2转化率为32.4%,CO选择性高达45.3%,但C2+OH选择性却只有11.8%。相比之下,在CO2加氢反应中表现出弱RWGS活性的Co基催化剂[5, 16, 17]引起研究者的关注。但因其优异的加氢性能,Co基催化剂通常被运用于CO2甲烷化反应[18, 19]。如1%Na/20%Co-SiO2催化剂[13],在H2预还原后主要产物是CH4(选择性80.0%)和C2-4烃。因此,本研究的研究思路在于降低Co基催化剂加氢性能,提高醇类产物的选择性。
本研究设计了一种以钙钛矿型复合氧化物(PTO)LaCo1-xGaxO3为前驱体的新催化剂,还原后得到的Co基催化剂用于CO2加氢直接制EtOH的反应。实验发现,LaCo0.7Ga0.3O3前驱体还原得到的Co/La2O3-La4Ga2O9催化剂表现出很高的EtOH选择性和良好的活性;催化剂中Ga对CO2加氢制EtOH的过程起到促进作用。通过表征,研究了催化剂的结构组成与催化性能的关系,推测出了CO2加氢合成EtOH的反应机理。
La(NO3)3·6H2O, Co(NO3)2·6H2O,柠檬酸,分析纯(AR), 阿拉丁试剂有限公司;Ga(NO3)3·9H2O,AR,北京华威锐科化工有限公司;乙二醇,无水乙醇,AR,天津光复化工研究所;去离子水,天津大学;二氧化碳、氢气和氮气混合气,99.999%,天津市液化有限公司。
采用柠檬酸络合法制备一系列以PTO为前驱体的LaCo1-xGaxO3 (x=0、0.2、0.3、0.4和0.5)。首先按照物质的量比La:Co:Ga=1:(1-x):x取一定量的La(NO3)3·6H2O,Co(NO3)2·6H2O和Ga(NO3)3·9H2O与柠檬酸和乙二醇一并加入去离子水中溶解,其中,柠檬酸物质的量为总阳离子量的1.2倍,乙二醇物质的量为柠檬酸的0.2倍。所得混合溶液放入水浴锅80 ℃搅拌蒸干后,烘箱120 ℃干燥12 h,得到蓬松的样品。然后以2 ℃/min的速率程序升温至350 ℃煅烧2 h,再升温至650 ℃煅烧5 h,得到前驱体LaCo1-xGaxO3 (x=0、0.2、0.3、0.4和0.5)。然后在选定温度下还原3 h, 得到催化剂,升温速率设定为2 ℃/min,由H2-TPR测试结果确定钴离子被完全还原的温度,还原温度分别取660、740、740、750和780 ℃。
X射线粉末衍射(XRD)表征选用Bruker D8-Focus型Cu靶X射线衍射仪测试,测试条件:λ=0.15406 nm,20°-80°扫速,扫描速率8(°)/min。
选用Agilent 7700X型电感耦合等离子体发射光谱仪测定催化剂中的钴镓比。测试准备:将催化剂溶解到强酸中配置成溶液,然后稀释到指定浓度。
N2吸附-脱附测试在Quantachrome Quadra Sorb SI型孔径分布测试仪进行,用BET方程计算比表面积,BJH法计算孔容。
程序升温脱附(H2/CO2-TPD)表征选用天津大学北洋化工实验设备公司自主研制的吸附-脱附还原反应多功能反应器测试。测试步骤如下:100 mg试样还原后,先用氢气或氦气(30 mL/min) 300 ℃原位预加热1 h,然后用氦气吹扫30 min,随后催化剂在氦气中冷却到50 ℃,切换阀门通入H2/CO2吸附气体2 h,再通入氦气吹扫30 min,待检测器基线稳定后,在氦气吹扫升温的过程中记录H2/CO2探针分子脱附曲线。测试条件:测试温度40-900 ℃,升温速率10 ℃/min。催化剂表面Co的分散度可基于H2-TPD数据通过下列等式计算:
|
$ D(\% ) = \frac{{{N_s}}}{{{N_A}}} \times 100\% $ |
(1) |
式中, Ns:催化剂表面Co原子的数目;NA:催化剂总的Co原子的总数目;假设H/Co=1[20, 21]。
场发射透射电子显微镜(TEM)表征选用JEOL JEM-2100F电子显微镜测试。测试准备:样品用无水乙醇稀释、研磨、静置后,取上层清液用无水乙醇稀释,超声20 min后滴加至铜网上,待样品自然干燥后测试。
X射线光电子能谱(XPS)表征选用美国Thermo ESCALAB 250Xi测试仪测试。测试条件:单色Al Kα(hv=1486.6 eV),功率150 W,500 μm束斑。电子结合能用样品C 1s峰(284.8 eV)作为内标校正。
热重(TG)分析选用DTG-50/50H热分析仪测试,计算催化剂上的焦炭和炭的沉积量。测试条件:空气气氛,升温速率10 ℃/min,测试20-900 ℃,20 mg催化剂。炭沉积量由公式(2)计算:
|
$ 炭沉积量=\frac{{{W_1} - {W_2}}}{{{W_1}}} \times 100\% $ |
(2) |
式中, W1和W2分别为TG测试前后催化剂的质量。
在微型固定床反应装置上评价催化剂催化CO2加氢制EtOH的活性。将0.5 g催化剂(40-60目)与石英砂等体积混合均匀后装入反应管,通入反应气体并升压,升温到所需的反应温度,维持12 h达到稳定状态后测试。测试条件:混合气H2/CO2=3.0,3 MPa,空速为3000 mL/(gcat·h),反应通入N2作为内标气体测试产物气体组成。采用SP-3400型气相色谱(GC)在线检测产物组成。气相产物(CO、CO2、H2、CH4)由TCD检测,碳氢化合物和液相产物由FID检测。其中,CO2转化率(xCO2),产物选择性(si)和总醇中各种醇的质量分数(wi)计算公式如下:
|
$ {x_{{\text{C}}{{\text{O}}_2}}} = \frac{{{{\left[ {{\text{C}}{{\text{O}}_2}} \right]}_{{\text{in}}}} - {{\left[ {{\text{C}}{{\text{O}}_2}} \right]}_{{\text{out}}}} }}{{{{[{\text{CO}}]}_{{\text{in}}}}}} \times 100\% $ |
(3) |
|
$ {s_i} = \frac{{n{{\text{C}}_i}}}{{\sum n {C_i}}} \times 100\% $ |
(4) |
|
$ {w_i} = \frac{{{m_i}}}{{\sum {{m_i}} }} \times 100\% $ |
(5) |
式中, [CO2]in和[CO2]out分别为反应进气和出气中的CO2摩尔量;n和Ci分别是含碳产物i的碳原子数和摩尔数;mi为液相产物中各个特定醇的质量。
图 1(a)为不同钴镓比LaCo1-xGaxO3前驱体煅烧后的XRD表征。由图 1(a)可知,对比LaCoO3(PDF#75-0279)的标准衍射峰,可以发现,所有样品的XRD谱图中只检测到归属于LaCo1-xGaxO3的衍射峰,未发现其他杂相的衍射峰,表明镓离子成功掺入LaCoO3中形成镧钴镓PTO。这保证了Co和Ga离子在原子水平上均匀混合,两者紧密接触形成相互作用。如图 1(a)插图所示,随着掺杂Ga含量的增多,样品的XRD衍射峰向小角度逐渐偏移。这是由于Co3+(0.0545 nm)和Ga3+(0.0620 nm)的不同造成的,离子半径大的Ga3+掺杂到LaCoO3中形成LaCo1-xGaxO3钙钛矿会导致晶格间距变化,且随着Ga掺杂量的增多,晶格间距的差距逐渐增大,表现为XRD衍射峰向低角度偏移。这表明镓元素成功进入LaCoO3钙钛矿晶格中。
图 1(b)为还原后的LaCo1-xGaxO3催化剂(x=0、0.2、0.3、0.4和0.5)的XRD谱图。由图 1(b)可知,谱图中归属于钙钛矿的衍射峰消失,这说明钙钛矿结构被破坏。当x=0时,出现归属于Co(111)和(100)晶面的特征衍射峰(PDF#88-2325),以及La2O3(101)、(102)和(110)晶面的特征衍射峰(PDF#74-2430)。当x=0.2、0.3和0.4时,出现对应于Co(PDF#88-2325)、La2O3(PDF#74-2430)以及La4Ga2O9(PDF#53-1108)的特征峰。而当x=0.5时,La2O3的特征峰消失,只有Co(PDF#88-2325)和La4Ga2O9(PDF#53-1108)的衍射峰出现,这符合化学计量比的规律。观察发现,随Ga含量的增加,La4Ga2O9衍射峰强度逐渐增强,而La2O3衍射峰的峰强度逐渐减弱。
图 2为前驱体为LaCo1-xGaxO3的催化剂(x=0和0.3)在煅烧后、还原后以及在给定的反应条件下反应48 h后(当x=0.3时还含有进行100 h稳定性测试后的样品)的XRD表征结果。当x=0时,煅烧后形成单一的PTO,还原后钙钛矿结构被破坏,出现Co和La2O3的衍射峰。反应48 h后的样品XRD谱图中出现LaCO3OH(PDF#26-0815)和Co2C(PDF#72-1369)的衍射峰,而Co和La2O3的衍射峰消失。其中,LaCO3OH是由La2O3和CO2以及反应生成的H2O反应形成(La2O3+CO2→La2O2CO3, La2O2CO3+CO2+H2O→LaCO3OH[22]),Co2C的形成归因于金属钴在含CO2的反应气中被碳化[23]。类似的,当x=0.3时,煅烧后只检测到LaCo0.7Ga0.3O3的衍射峰,还原后的衍射峰归属于Co、La2O3和La4Ga2O9的存在。比较还原后,反应48 h以及100 h后的样品的XRD谱图可知,反应前后样品的XRD衍射峰强度无明显的变化。因此,可以推断出以LaCo0.7Ga0.3O3为前驱体的催化剂在反应过程中展现出很好的抗烧结能力。

对于以LaCo0.7Ga0.3O3为前驱体的催化剂有两点需要强调:第一点,比较H2-TPD和TEM以及XRD表征结果,发现反应前后(甚至是反应100 h以后)的催化剂的Co颗粒平均粒径相差不大,这说明以LaCo0.7Ga0.3O3为前驱体的催化剂有很好的抗烧结能力。第二点,与LaCoO3比较发现,即使是反应100 h后的LaCo0.7Ga0.3O3催化剂的XRD谱图上也没有发现与Co2C有关的衍射峰,这可能是因为Ga的掺杂有助于Co颗粒的分散[24]和抑制Co2C的形成。Du等[25]发现La2O3的添加可以促进Co2C的生成,而Al2O3的添加则会抑制Co2C的生成。基于Ga和Al在元素周期表上位于同一主族,可以推测Ga和Al对于抑制Co2C生成方面有类似的性质。
图 3为LaCo1-xGaxO3(x=0、0.2、0.3、0.4和0.5)的H2-TPR表征结果。由图 3可知,所有样品均出现两个明显的H2还原峰,对应PTO中的B位钴离子的两步还原过程。当x=0时,位于350-550 ℃的低温H2还原峰(峰α)归因于PTO晶格中的Co3+还原为Co2+的过程[26, 27]。
|
$ {\text{LaCo}}{{\text{O}}_3} + 1/2\ {{\text{H}}_2} \to {\text{LaCo}}{{\text{O}}_{2.5}} + 1/2\ {{\text{H}}_2}{\text{O}} $ |
(6) |

出现在550-750℃的H2还原峰(峰β)归因于Co2+进一步还原变成金属Co的过程[26, 27]。峰β分裂为两个峰,一般认为对应表面和体相中的Co2+还原为Co的过程[28],还原后形成Co/La2O3。
|
$ {\text{LaCo}}{{\text{O}}_{2.5}} + {{\text{H}}_2} \to 1/2{\text{L}}{{\text{a}}_2}{{\text{O}}_3} + {\text{Co}} + {{\text{H}}_2}{\text{O}} $ |
(7) |
当x=0.2-0.5时,与LaCoO3的H2-TPR谱图相比,可以看到峰α的还原温度向较低温度方向移动,峰β的还原温度向高温方向移动,这说明Ga的掺杂使PTO晶格中的Co3+还原成Co2+的过程变得相对容易,但对Co2+→Co的过程起到抑制作用。
H2-TPR的耗氢量表明还原至β峰对应的温度后,钴离子被完全还原为金属钴。所有样品的峰β和峰α的耗氢峰面积比接近2:1,与钴离子两步还原过程一致。对应数据见表 1。
 下载:
导出CSV
下载:
导出CSV
| Sample | Co loading/% | Co/Gaa | Co/Gab | Crystal size of Coc, d/nm | H2 uptake/ (mmol·g-1) |
Co dispersion/%c, e | H2 consumptions from TPR resultsf, g | Total theoretic H2consumptionsg | Reducibility degree of Con+/% | |
| α | β | |||||||||
| x=0 | 26.6 | - | - | 18.5 | 0.104 | 5.2 | 0.101 | 0.204 | 0.305 | 100 |
| x=0.2 | 20.6 | 4.00 | 3.97 | 8.8 | 0.184 | 11.5 | 0.077 | 0.162 | 0.242 | 98.8 |
| x=0.3 | 17.8 | 2.33 | 2.36 | 7.6(10.5)h | 0.188(0.137) | 13.4(9.8)h | 0.068 | 0.138 | 0.211 | 97.6 |
| x=0.4 | 15.0 | 1.50 | 1.50 | 6.9 | 0.173 | 14.4 | 0.064 | 0.122 | 0.180 | 103.3 |
| x=0.5 | 12.3 | 1.00 | 1.01 | 7.4 | 0.136 | 13.6 | 0.051 | 0.108 | 0.149 | 106.7 |
| a: Co/Ga ratio in synthesis system; b: Co/Ga ratio in sample measured from ICP; c: calculated from the results of H2-TPD; d: the crystal size for catalysts after reduction; e: assuming H/Co=1; f: experimental H2 consumptions calculated from TPR results using CuO as the reference material; g: the unit is mmol H2 per 50 mg of catalyst; h: crystal size of Co after 100 h stability test in the parentheses | ||||||||||
各样品的N2吸附-脱附等温线见图 4。
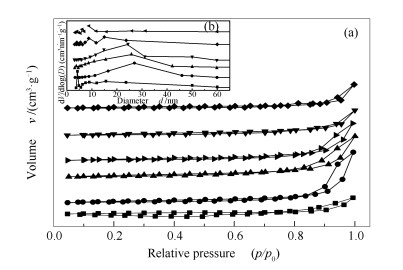
由图 4可知,所有样品的吸附等温线形态与Ⅳ型吸附等温线非常相似,并出现较弱的H3型滞留环,这说明样品中存在一定量的介孔(孔径在2-50 nm),这与BJH孔径分布曲线表征结果一致。观察到滞留环的起始位置在p/p0=0.8,这说明形成的孔大多是堆积孔[29],其原因可能是煅烧样品分解柠檬酸盐形成PTO结构的过程中,催化剂颗粒堆积形成多孔结构[30]。并且由表 2的数据可知,与LaCoO3相比,掺杂Ga后样品的比表面积和平均孔径增大,但是随着Ga掺杂量的增加,样品的比表面积又从9.6 m2/g减少到6.5 m2/g,平均孔径从27.0 nm减少至6.2 nm。结合图 1(b)的XRD结果分析,La4Ga2O9晶粒粒径随着Ga掺杂量的增加而逐渐增大(表现为La4Ga2O9的衍射峰强度逐渐增强)是催化剂比表面积和平均孔径逐渐减少的主要原因。
下载:
导出CSV
| Sample | Specific surface area A/(m2·g-1) | Total pore volume v/(cm3·g-1) | Average pore size d/nm |
| x=0 | 5.6 | 0.02 | 4.2 |
| x=0.2 | 9.6 | 0.04 | 27.0 |
| x=0.3 | 9.4(9.1)a | 0.02(0.03)a | 25.8(24.4)a |
| x=0.4 | 8.8 | 0.02 | 8.9 |
| x=0.5 | 6.5 | 0.02 | 6.2 |
| a: data in the parentheses are for the spent catalyst after 100 h reaction test | |||
对于LaCo0.7Ga0.3O3,反应100 h后,催化剂的比表面积由9.4 m2/g减少至9.1 m2/g,平均孔径由25.8 nm变为24.4 nm;同时,反应前后的N2吸附-脱附等温线和BJH孔径分布曲线的变化几乎可以忽略不计;均体现催化剂的抗烧结能力强。
不同钴镓比催化剂的CO2-TPD表征结果见图 5,记脱附峰的峰强最大的温度值为tm。tm位于100-200 ℃的脱附峰主要为吸附在弱碱性位上的CO2脱附,高温段(>600 ℃)出现的脱附峰主要是因为LaCO3OH的分解[22]和CO2与强碱性位之间的强作用力[31],由于CO2自身的低能级轨道和高电子亲和能(38 eV),CO2得电子的能力强,所以金属Co可以通过转移电子来吸附活化CO2[18, 19, 32],而Yazdani等[33]也指出,碱性氧化物La2O3上弱、中等和强碱性位均存在。所以位于200-500 ℃的脱附峰(峰α)主要为吸附在金属Co和La2O3上的CO2脱附,对应催化剂表面的中等碱性位[31]。一般认为,中等吸附强度的CO2(峰α)属于有效的活化,参与化学反应[31, 34]。

a: x=0; b: x=0.2, c: x=0.3, d: x=0.4; e: x=0.5
如上所述,峰α对应的CO2吸附应该是金属Co和La2O3共同作用的结果。虽然随着x的增加,催化剂中的Co和La2O3含量均逐渐减少,但是两者的分散度也都随之增加,在这两个因素的共同作用下催化剂对CO2吸附、活化效果在x=0.3时达到最佳状态,表现为峰α的峰面积达到最大值。而当x=0.5时,峰α的峰面积显著减少,说明当Ga的掺杂量过多时,催化剂表面的中等碱性位点数量反而减少。此时还原后催化剂中无La2O3的存在,结合XRD数据认为,还原后催化剂中La2O3含量影响催化剂吸附活化CO2的性能。通过计算,峰α所代表的化学吸附CO2的量占总吸附量的百分比顺序为:LaCo0.7Ga0.3O3(31.9%)> LaCo0.6Ga0.4O3(29.3%)> LaCo0.8Ga0.2O3(10.4%)> LaCoO3(7.9%)> LaCo0.5Ga0.5O3(3.4%)。表明前驱体为LaCo0.7Ga0.3O3的催化剂反应性能最好,这与活性测试结果相符。
采用XPS表征来进一步探究催化剂的表面状态。其中,催化剂表面各元素的含量见表 3。图 6为LaCoO3和LaCo0.7Ga0.3O3催化剂反应前后的XPS谱图及其拟合结果。由图 6(a)可知,反应后的LaCoO3催化剂的La 3d谱图在835.2和852.0 eV处出现分别归属于La 3d5/2和La 3d3/2的特征峰,表明反应后LaCO3OH的存在[35],与XRD表征结果一致。与反应后的LaCoO3的La 3d峰相比,反应后的LaCo0.7Ga0.3O3催化剂的La 3d5/2电子结合能(834.7 eV)和La 3d3/2电子结合能(851.5 eV)都向低结合能进行了位移,这应该是因为催化剂中部分La以La4Ga2O9形式存在。由图 6(b)可知,反应前后LaCo0.7Ga0.3O3催化剂均出现在17.0和19.0 eV附近的特征峰对应Ga3+的电子结合能[36],结合XRD谱图证明反应前后催化剂中La4Ga2O9的存在。由图 6(c)可知,对Co 2p谱峰进行分峰处理。778.4 eV附近的特征峰对应Co0的电子结合能,780.1和795.7 eV附近出现的特征峰以及相邻的卫星峰(satellite peak)分别对应于Co2+的Co 2p3/2和Co 2p1/2的电子结合能。表明反应后的LaCoO3和LaCo0.7Ga0.3O3催化剂表面均存在金属Co和Co2+。结合XRD表征结果,LaCoO3中的Co2+可以归因于反应过程中生成的Co2C;而LaCo0.7Ga0.3O3的Co2+可能是因为Co0在反应过程中或催化剂暴露于空气被氧化所致。与标准Co0电子结合能(777.9 eV)[37, 38]和标准Ga3+电子结合能(17.4 eV)[36]相比,LaCo0.7Ga0.3O3催化剂中Co0电子结合能升高,Ga3+电子结合能降低。这可能是因为Ga离子与金属Co之间存在电子协同作用,含Ga化合物的形成使部分Co处于带正电状态。观察Co 2p峰的峰位置和形状,与反应后的LaCoO3催化剂相比,反应后的LaCo0.7Ga0.3O3的Co 2p的特征峰的形状和位置与金属Co的Co 2p1/2和Co 2p3/2峰很接近,且在主峰临近高结合能一侧的卫星峰的峰强小,这说明反应过程中LaCo0.7Ga0.3O3催化剂表面的Co0物种部分被氧化,催化剂是以金属Co和CoO的混合状态存在的[39],但XRD谱图中并未检测到CoO的衍射峰,这可能是因为反应过程Co颗粒氧化形成的CoO颗粒粒径小,分散度高的缘故。
下载:
导出CSV
| Sample | Atomic ratio/% | ||
| La/Σ[M]a | Co/Σ[M]a | Ga/Σ[M]a | |
| LaCoO3-used | 56.1(50.0)b | 43.9(50.0)b | - |
| LaCo0.7Ga0.3O3-used | 47.0(50.0)b | 15.5(35.0)b | 37.5(15.0)b |
| a: the atomic ratio of metal M is M/(La+Co+Ga) and the data in the parentheses are theoretical values | |||

由表 3可知,掺杂Ga后,催化剂表面Co/Σ[M]物质的量比明显低于体相,而Ga存在表面富集现象。这可能是因为含Ga的化合物(La4Ga2O9)与Co之间存在强相互作用,促进La4Ga2O9迁移到催化剂表面,表现为Ga元素的表面富集。
图 7为LaCo0.7Ga0.3O3催化剂反应前后的TEM照片。图 7(a)中可识别的较暗区域以及黑点归属于Co颗粒,还原后的催化剂表面活性组分Co颗粒分布比较均匀,Co颗粒的晶粒粒径为3.4-15.0 nm,平均晶粒粒径为7.7 nm,与H2-TPD表征计算结果相符。图 7(a′)中高倍透射电镜照片中的晶格条纹分别对应金属Co的(100)晶面(晶面间距d=0.217 nm),La4Ga2O9的(022)以及(-221)晶面(晶面间距分别为0.391和0.324 nm),说明活性组分Co均匀地负载在载体氧化物上。类似的,图 7(b)为反应100 h后的催化剂的TEM照片,其中,Co颗粒的晶粒粒径为4.3-19.9 nm,平均晶粒粒径为10.8 nm。对比图 7(a)和图 7(b)可知,反应后催化剂的活性组分发生轻微团聚烧结现象,在TEM照片上显示出不同的衬度,聚集体尺寸稍微变大。图 7(b′)显示的晶格条纹与CoO(101)晶面(晶面间距d=0.245 nm)和La4Ga2O9(002)晶面(晶面间距d=0.547 nm)一致,而图 7(b″)中晶面间距分别为0.205和0.324 nm的晶格条纹分别对应金属Co(111)晶面和La4Ga2O9(-221)晶面,与XPS表征结果一致。反应前后TEM表征结果对比再次证实反应气氛中的CO2或/和过程中产生的水使得部分Co颗粒被氧化成CoO。

图 8为反应后催化剂的TG曲线。由图 8可知,反应48 h的LaCoO3催化剂的TG曲线出现两处明显的失重峰,分别归属于大分子烃类和石墨化炭的燃烧[40],总失重率约为15.4%,而相同反应条件下,反应48和100 h的LaCo0.7Ga0.3O3催化剂的TG曲线上只有一个归属于大分子烃类和无定型炭燃烧的失重峰,总失重率分别为7.8%和9.9%。所有样品在150-350 ℃均存在增重的现象,可归属于催化剂中的金属Co被空气气氛中氧气氧化的过程[41]。计算反应48 h的催化剂单位质量Co对应增质量,相比于反应后的LaCoO3(单位质量Co增重0.11 g),LaCo0.7Ga0.3O3在反应后的氧化增重现象并不明显(单位质量Co增质量0.02 g),这可能是因为反应过程中LaCo0.7Ga0.3O3中部分金属Co已经被氧化,结合性能测试结果,LaCo0.7Ga0.3O3催化剂反应过程中部分金属Co被氧化可能是液相分布不稳定的一个原因。

TG表征结果表明,Ga的掺杂提高了催化剂的抗积炭能力,这可能是因为两方面的原因,第一,氧空位的消积炭作用[42]:反应气氛中H2的还原作用促进La4Ga2O9表面氧空位的形成,在氧空位上吸附、活化的氧物种可以起到消积炭的作用;第二,Co2C是金属Co表面形成石墨化炭的中间物种[43]。如前所述,反应后的LaCoO3催化剂上存在Co2C物种,同时热分析结果显示反应过程中LaCoO3催化剂上形成石墨化炭;而XRD表征和热分析结果都表明反应后的LaCo0.7Ga0.3O3催化剂中检测不到Co2C,也无石墨化炭形成;这些结果相互印证。
表 4为以LaCo1-xGaxO3为前驱体的催化剂催化CO2加氢制EtOH的CO2转化率和产物选择性。反应条件为:t=240 ℃,p=3MPa,n(H2)/n(CO2)=3.0,GSHV=3000 mL/(gcat·h),反应时间为18 h。由表 4可知,随着镓含量的增加,CO2的转化率和CH4的选择性持续降低,总醇选择性持续升高,而总醇产物中的EtOH占比表现出先升高后降低的趋势,当x=0.3时达到最大。
下载:
导出CSV
| Sample | CO2 conversion x/% | Selectivityb sC, mol/% | Alcohol distributionc w/% | ||||
| CH4 | C2+H | ROH | MeOH | EtOH | |||
| x=0 | 30.4 | 97.8 | 1.7 | 0.5 | 77.0 | 23.0 | |
| x=0.2 | 10.6 | 37.4 | 2.5 | 60.1 | 30.2 | 69.8 | |
| x=0.3 | 9.8 | 23.1 | 2.2 | 74.7 | 11.9 | 88.1 | |
| x=0.4 | 8.1 | 12.2 | 2.1 | 85.7 | 27.5 | 72.5 | |
| x=0.5 | 4.78 | 11.1 | 2.4 | 86.5 | - | - | |
|
a: reaction conditions: 240 ℃, 3 MPa, n(H2)/n(CO2) = 3.0, GSHV = 3000 mL/(gcat·h); the data were obtained after 18 h on stream; b: product selectivity was based carbon molar quantity, defined as the carbon molar quantity in a carbon-containing product divided by converted carbon moles; C2+H represents hydrocarbons exclusive of methane; c: alcohol distribution (mass ratio) is the weight fraction of each alcohol in total alcohols |
|||||||
其中,当x=0时,催化剂的CO2转化率达到30.4%,但CH4的选择性也高达97.8%,表现出Co基催化剂优异的CO2甲烷化的性能[18, 19],而少量醇的形成可以归因于Co和Co2C的协同催化,这个已有报道[44]。当x=0.2-0.5时,LaCo0.7Ga0.3O3催化剂表现出良好的催化CO2加氢制EtOH的性能,当CO2转化率为9.8%时,总醇选择性达到74.7%,液相产物中的EtOH质量分数可达到88.1%。表 5列出的是文献报道的一些用于CO2加氢反应的Co基催化剂[11, 18, 45]。由表 5可以看出,本研究高的EtOH选择性在已有的文献报道中难以找到。
下载:
导出CSV
结合XPS表征合理推断:Ga的掺杂影响催化剂的催化性能,使得CO2转化率降低;这是因为Ga的掺杂抑制催化剂表面Co元素的富集,催化剂表面的Co活性位点减少,并且Ga掺杂量的增加对应着催化剂中的Co含量降低(性能测试时,固定LaCo1-xGaxO3催化剂的总用量)。而CH4选择性降低,总醇产物中的EtOH占比增加,则是因为Co与Ga之间的相互作用使得催化剂界面处的部分Co以Coδ+的状态存在,Co和Coδ+的协同作用可以适当减弱金属Co的加氢性能,抑制CO2加氢生成CH4,并且促进EtOH的生成[46, 47]。
图 9为反应温度对LaCo1-xGaxO3催化剂(x=0、0.2-0.5)催化性能的影响。由图 9可知,反应温度对LaCo1-xGaxO3催化剂的催化性能影响较大。相对于LaCoO3催化剂,LaCo1-xGaxO3催化剂(0.2-0.5)在整个反应温度区间(230-290 ℃)均呈现出高总醇选择性和高EtOH分布。具体来说,随着反应温度的升高,LaCo1-xGaxO3催化剂的CO2转化率升高(图 9(a)),总醇选择性(图 9(b))和液相产物中EtOH含量占比(图 9(c))下降。而x=0.5时,虽然在230-270 ℃反应区间的总醇选择性下降不明显,但是CO2转化率一直保持在较低的水平(5%左右)。综合CO2转化率和EtOH选择性这两个评价催化剂性能的重要指标,选择240 ℃为最佳反应温度。

图 10为液相产物分布随反应时间的变化。

由图 10可知,随着反应的进行,液相产物中的MeOH质量分数逐渐增加,而EtOH质量分数相应减少。据文献报道[7],在CO2加氢制EtOH的反应中,Co基催化剂中的Co和CoO的相对含量影响液相产物中醇类分布。催化剂中的CoO含量的增加促进液相产物中的MeOH的生成。基于以上表征结果与文献阐述,本研究认为随着反应的进行,反应气氛中的CO2或/和水会使催化剂中的金属Co活性组分被部分氧化成CoO[48, 49],导致液相醇分布发生变化。
本研究提出初步机理:CO2和H2在Co/La2O3-La4Ga2O9催化剂上发生催化反应形成EtOH的过程是金属Co-Coδ+协同催化生成。
金属Co解离H2形成H*[11, 18];金属Co解离吸附CO2,随后形成CH3*[7];Coδ+上发生CO2的非解离吸附,并接受金属Co上迁移来的H*形成HCOO*物种。随后分解形成CO*[18, 19];CH3*与相邻的CO*耦合形成CH3CO*,加氢形成EtOH物质。
催化剂表面金属Co活性位点上形成的CH3*物种可以促进HCOO*物种转化为CH3CO*中间体,利于EtOH的生成。Coδ+是CO2非解离吸附的活性位,含Ga化合物La4Ga2O9起稳定Coδ+的作用。
本研究采用柠檬酸络合法合成一系列以LaCo1-xGaxO3(x=0、0.2-0.5)为前驱体的Co基催化剂,将其用于CO2加氢直接制EtOH反应。实验结果表明,相比于Co/La2O3,Co/La2O3-La4Ga2O9表现出优良的催化性能(适宜的CO2转化率,显著高的EtOH选择性,抗积炭能力强),其中,x=0.3时的催化剂表现出最佳的催化性能。表征结果表明,Ga的掺杂不仅可以提高催化剂表面活性组分金属Co的分散度,还通过形成含Ga化合物(La4Ga2O9)与金属Co产生相互作用使得界面产生Coδ+,Co和Coδ+协同作用促进EtOH的生成。
WANG D, BI Q, YIN G, ZHAO W, HUANG F, XIE X, JIANG M. Direct synthesis of ethanol via CO2 hydrogenation using supported gold catalysts[J]. Chem Commun (Camb), 2016, 52(99): 14226-14229. doi: 10.1039/C6CC08161D
WANG W, WANG S, MA X, GONG J. Recent advances in catalytic hydrogenation of carbon dioxide[J]. Chem Soc Rev, 2011, 40(7): 3703-3727. doi: 10.1039/c1cs15008a
WEI W, JINLONG G. Methanation of carbon dioxide: An overview[J]. Front Chem Sci Eng, 2011, 5(1): 2-10. doi: 10.1007/s11705-010-0528-3
AZIZ M A A, JALIL A A, TRIWAHYONO S, AHMAD A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects[J]. Green Chem, 2015, 17(5): 2647-2663. doi: 10.1039/C5GC00119F
POROSOFF M D, YAN B, CHEN J G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities[J]. Energy Environ Sci, 2016, 9(1): 62-73. doi: 10.1039/C5EE02657A
SUN K, LU W, QIU F, LIU S, XU X. Direct synthesis of DME over bifunctional catalyst: Surface properties and catalytic performance[J]. Appl Catal A: Gen, 2003, 252(2): 243-249. doi: 10.1016/S0926-860X(03)00466-6
WANG L, WANG L, ZHANG J, LIU X, WANG H, ZHANG W, YANG Q, MA J, DONG X, YOO S J, KIM J, MENG X, XIAO F. Selective hydrogenation of CO2 to ethanol over cobalt catalysts[J]. Angew Chem Int Ed, 2018, 57(21): 6104-6108. doi: 10.1002/anie.201800729
WANG D, BI Q, YIN G, ZHAO W, HUANG F, XIE X, JIANG M. Direct synthesis of ethanol via CO2 hydrogenation using supported gold catalysts[J]. Chem Commun, 2016, 52(99): 14226-14229. doi: 10.1039/C6CC08161D
QIAN Q, CUI M, HE Z, WU C, ZHU Q, ZHANG Z, MA J, YANG G, ZHANG J, HAN B. Highly selective hydrogenation of CO2 into C2+ alcohols by homogeneous catalysis[J]. Chem Sci, 2015, 6(10): 5685-5689. doi: 10.1039/C5SC02000J
HE Z, QIAN Q, MA J, MENG Q, ZHOU H, SONG J, LIU Z, HAN B. Water-enhanced synthesis of higher alcohols from CO2 Hydrogenation over a Pt/Co3O4 catalyst under milder conditions[J]. Angew Chem Int Ed, 2016, 55(2): 737-741. doi: 10.1002/anie.201507585
OUYANG B, XIONG S, ZHANG Y, LIU B, LI J. The study of morphology effect of Pt/Co3O4 catalysts for higher alcohol synthesis from CO2 hydrogenation[J]. Appl Catal A: Gen, 2017, 543: 189-195. doi: 10.1016/j.apcata.2017.06.031
PRIETO G. Carbon dioxide hydrogenation into higher hydrocarbons and oxygenates: Thermodynamic and kinetic bounds and progress with heterogeneousand homogeneous catalysis[J]. ChemSusChem, 2017, 10(6): 1056-1070. doi: 10.1002/cssc.201601591
GNANAMANI M K, JACOBS G, KEOGH R A, SHAFER W D, SPARKS D E, HOPPS S D, THOMAS G A, DAVIS B H. Fischer-Tropsch synthesis: Effect of pretreatment conditions of cobalt on activity and selectivity for hydrogenation of carbon dioxide[J]. Appl Catal A: Gen, 2015, 499: 39-46. doi: 10.1016/j.apcata.2015.03.046
GNANAMANI M K, HAMDEH H H, JACOBS G, SHAFER W D, HOPPS S D, THOMAS G A, DAVIS B H. Hydrogenation of carbon dioxide over K-promoted FeCo bimetallic catalysts prepared from mixed metal oxalates[J]. ChemCatChem, 2017, 9(7): 1303-1312. doi: 10.1002/cctc.201601337
GUO H, LI S, PENG F, ZHANG H, XIONG L, HUANG C, WANG C, CHEN X. Roles investigation of promoters in K/Cu-Zn catalyst and higher alcohols synthesis from CO2 hydrogenation over a novel two-stage bed catalyst combination system[J]. Catal Lett, 2015, 145(2): 620-630. doi: 10.1007/s10562-014-1446-7
GNANAMANI M K, JACOBS G, HAMDEH H H, SHAFER W D, LIU F, HOPPS S D, THOMAS G A, DAVIS B H. Hydrogenation of carbon dioxide over Co-Fe bimetallic catalysts[J]. ACS Catal, 2016, 6(2): 913-927. doi: 10.1021/acscatal.5b01346
POUR A N, HOSAINI E, IZADYAR M, HOUSAINDOKHT M R. Particle size effects in Fischer-Tropsch synthesis by Co catalyst supported on carbon nanotubes[J]. Chin J Catal, 2015, 36(8): 1372-1378. doi: 10.1016/S1872-2067(15)60840-3
ZHOU G, LIU H, XING Y, XU S, XIE H, XIONG K. CO2 hydrogenation to methane over mesoporous Co/SiO2 catalysts: Effect of structure[J]. J CO2 Util, 2018, 26: 221-229. doi: 10.1016/j.jcou.2018.04.023
LIU H, XU S, ZHOU G, XIONG K, JIAO Z, WANG S. CO2 hydrogenation to methane over Co/KIT-6 catalysts: Effect of Co content[J]. Fuel, 2018, 217: 570-576. doi: 10.1016/j.fuel.2017.12.112
LI Z, SI M, XIN L, LIU R, LIU R, LV J. Cobalt catalysts for Fischer-Tropsch synthesis: The effect of support, precipitant and pH value[J]. Chin J Chem Eng, 2018, 26(4): 747-752. doi: 10.1016/j.cjche.2017.11.001
JOHNSON G R, BELL A T. Effects of Lewis acidity of metal oxide promoters on the activity and selectivity of Co-based Fischer-Tropsch synthesis catalysts[J]. J Catal, 2016, 338: 250-264. doi: 10.1016/j.jcat.2016.03.022
LEE M, JUNG W. Hydrothermal synthesis of LaCO3OH and Ln3+-doped LaCO3OH powders under ambient pressure and their transformation to La2O2CO3 and La2O3[J]. Bull Korean Chem Soc, 2013, 34(12): 3609-3614. doi: 10.5012/bkcs.2013.34.12.3609
CHAKRABARTI D, DE KLERK A, PRASAD V, GNANAMANI M K, SHAFER W D, JACOBS G, SPARKS D E, DAVIS B H. Conversion of CO2 over a Co-based Fischer-Tropsch catalyst[J]. Ind Eng Chem Res, 2015, 54(4): 1189-1196. doi: 10.1021/ie503496m
GAO P, LI F, ZHAN H, ZHAO N, XIAO F, WEI W, ZHONG L, WANG H, SUN Y. Influence of Zr on the performance of Cu/Zn/Al/Zr catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol[J]. J Catal, 2013, 298: 51-60. doi: 10.1016/j.jcat.2012.10.030
DU H, ZHU H, ZHAO Z, DONG W, LUO W, LU W, JIANG M, LIU T, DING Y. Effects of impregnation strategy on structure and performance of bimetallic CoFe/AC catalysts for higher alcohols synthesis from syngas[J]. Appl Catal A: Gen, 2016, 523: 263-271. doi: 10.1016/j.apcata.2016.06.022
BEDEL L, ROGER A C, ESTOURNES C, KIENNEMANN A. Co0 from partial reduction of La(Co, Fe)O3 perovskites for Fischer-Tropsch synthesis[J]. Catal Today, 2003, 85(2/4): 207-218.
TONIOLO F S, MAGALHÃES R N S H, PEREZ C A C, SCHMAL M. Structural investigation of LaCoO3 and LaCoCuO3 perovskite-type oxides and the effect of Cu on coke deposition in the partial oxidation of methane[J]. Appl Catal B: Environ, 2012, 117/118: 156-166. doi: 10.1016/j.apcatb.2012.01.009
ONRUBIA J A, PEREDA-AYO B, DE-LA-TORRE U, GONZÁLEZ-VELASCO J R. Key factors in Sr-doped LaBO3 (B=Co or Mn) perovskites for NO oxidation in efficient diesel exhaust purification[J]. Appl Catal B: Environ, 2017, 213: 198-210. doi: 10.1016/j.apcatb.2017.04.068
NIU T, LIU G L, CHEN Y, YANG J, WU J, CAO Y, LIU Y. Hydrothermal synthesis of graphene-LaFeO3 composite supported with Cu-Co nanocatalyst for higher alcohol synthesis from syngas[J]. Appl Surf Sci, 2016, 364: 388-399. doi: 10.1016/j.apsusc.2015.12.164
GUO S, LI S, ZHONG H, GONG D, WANG J, KANG N, ZHANG L, LIU G, LIU Y. Mixed oxides confined and tailored cobalt nanocatalyst for direct ethanol synthesis from syngas: A catalyst designing by using Perovskite-Type oxide as the precursor[J]. Ind Eng Chem Res, 2018, 57(6): 2404-2415. doi: 10.1021/acs.iecr.7b04336
PODILA S, DRISS H, ZAMAN S F, ALHAMED Y A, ALZAHRANI A A, DAOUS M A, PETROV L A. Hydrogen generation by ammonia decomposition using Co/MgO-La2O3 catalyst: Influence of support calcination atmosphere[J]. J Mol Catal A: Chem, 2016, 414: 130-139. doi: 10.1016/j.molcata.2016.01.012
DE LA PEÑA O SHEA V A, GONZÁLEZ S, ILLAS F, FIERRO J L G. Evidence for spontaneous CO2 activation on cobalt surfaces[J]. Chem Phys Lett, 2008, 454(4/6): 262-268.
YAZDANI P, WANG B, GAO F, KAWI S, BORGNA A. Role of the strong lewis base sites on glucose hydrogenolysis[J]. ChemCatChem, 2018, 10(17): 3845-3853. doi: 10.1002/cctc.201800427
PODILA S, DRISS H, ZAMAN S F, ALI A M, AL-ZAHRANI A A, DAOUS M A, PETROV L A. Effect of preparation methods on the catalyst performance of Co/Mg La mixed oxide catalyst for COx-free hydrogen production by ammonia decomposition[J]. Int J Hydrogen Energy, 2017, 42(38): 24213-24221. doi: 10.1016/j.ijhydene.2017.07.112
ZHANG Y, HAN K, CHENG T, FANG Z. Synthesis, characterization, and photoluminescence property of LaCO3OH microspheres[J]. Inorg Chem, 2007, 46(11): 4713-4717. doi: 10.1021/ic0701458
JENA H, GOVINDAN KUTTY K V, KUTTY T R N. Novel wet chemical synthesis and ionic transport properties of LaGaO3 and selected doped compositions at elevated temperatures[J]. Mater Sci Eng B, 2004, 113(1): 30-41. doi: 10.1016/j.mseb.2004.06.017
JAIN R, GOPINATH C S. New strategy toward a dual functional nanocatalyst at ambient conditions: Influence of the Pd-Co interface in the catalytic activity of Pd@Co core-shell nanoparticles[J]. ACS Appl Mater Interfaces, 2018, 10(48): 41268-41278. doi: 10.1021/acsami.8b12940
BIESINGER M C, PAYNE B P, GROSVENOR A P, LAU L W M, GERSON A R, SMART R S C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni[J]. Appl Surf Sci, 2011, 257(7): 2717-2730. doi: 10.1016/j.apsusc.2010.10.051
LIU L F, KANG J F, WANG Y, TANG H, KONG L G, SUN L, ZHANG X, HAN R Q. The influence of hydrogen annealing on magnetism of Co-doped TiO2 films prepared by sol-gel method[J]. J Magn Magn Mater, 2007, 308(1): 85-89.
ZHAO L, HAN T, WANG H, ZHANG L, LIU Y. Ni-Co alloy catalyst from LaNi1-xCoxO3 perovskite supported on zirconia for steam reforming of ethanol[J]. Appl Catal B: Environ, 2016, 187: 19-29. doi: 10.1016/j.apcatb.2016.01.007
SAN-JOSÉ-ALONSO D, JUAN-JUAN J, ILLÁN-GÍMEZ M J, ROMÁN-MARTÍNEZ M C. Ni, Co and bimetallic Ni-Co catalysts for the dry reforming of methane[J]. Appl Catal A: Gen, 2009, 371(1/2): 54-59.
LIU F, ZHAO L, WANG H, BAI X, LIU Y. Study on the preparation of Ni-La-Ce oxide catalyst for steam reforming of ethanol[J]. Int J Hydrogen Energy, 2014, 39(20): 10454-10466. doi: 10.1016/j.ijhydene.2014.05.036
YANG Q, CAO A, KANG N, AN K, LIU Z, LIU Y. A new catalyst of Co/La2O3-doped La4Ga2O9 for direct ethanol synthesis from syngas[J]. Fuel Process Technol, 2018, 179: 42-52. doi: 10.1016/j.fuproc.2018.06.011
PEI Y, LIU J, ZHAO Y, DING Y, LIU T, DONG W, ZHU H, SU H, YAN L, LI J, LI W. High alcohols synthesis via Fischer-Tropsch reaction at cobalt metal/carbide interface[J]. ACS Catal, 2015, 5(6): 3620-3624. doi: 10.1021/acscatal.5b00791
R W DORNER D R H F, WILLAUER H D. Influence of gas feed composition and pressure on the catalytic conversion of CO2 to hydrocarbons using a traditional cobalt-based Fischer-Tropsch catalyst[J]. Energy Fuels, 2009, 23: 4190-4195. doi: 10.1021/ef900275m
JIAO G, DING Y, ZHU H, LI X, LI J, LIN R, DONG W, GONG L, PEI Y, LU Y. Effect of La2O3 doping on syntheses of C1-C18 mixed linear α-alcohols from syngas over the Co/AC catalysts[J]. Appl Catal A: Gen, 2009, 364(1/2): 137-142.
SMITH M L, KUMAR N, SPIVEY J J. CO adsorption behavior of Cu/SiO2, Co/SiO2, and CuCo/SiO2 catalysts studied by in situ DRIFTS[J]. J Phys Chem C, 2012, 116(14): 7931-7939. doi: 10.1021/jp301197s
ZHANG Y, JACOBS G, SPARKS D E, DRY M E, DAVIS B H. CO and CO2 hydrogenation study on supported cobalt Fischer-Tropsch synthesis catalysts[J]. Catal Today, 2002, 71(3/4): 411-418.
TAKANABE K, NAGAOKA K, NARIAI K, AIKA K. Titania-supported cobalt and nickel bimetallic catalysts for carbon dioxide reforming of methane[J]. J Catal, 2005, 232(2): 268-275. doi: 10.1016/j.jcat.2005.03.011

图 1 LaCo1-xGaxO3样品(x=0、0.2、0.3、0.4和0.5)的XRD谱图
Figure 1 XRD patterns of the LaCo1-xGaxO3 samples (x=0, 0.2, 0.3, 0.4 and 0.5)

图 2 LaCoO3(a)和LaCo0.7Ga0.3O3 (b)样品煅烧后、还原后和反应后的XRD谱图
Figure 2 XRD patterns of the fresh, reduced and spent LaCoO3 (a) and LaCo0.7Ga0.3O3 (b) samples

图 3 LaCo1-xGaxO3样品(x=0,0.2,0.3,0.4和0.5)的H2-TPR谱图
Figure 3 H2-TPR profiles of the LaCo1-xGaxO3 samples (x=0, 0.2, 0.3, 0.4 and 0.5)

图 4 还原后的LaCoO3(■)、LaCo0.8Ga0.2O3(●)、LaCo0.7Ga0.3O3(▲)以及LaCo0.7Ga0.3O3反应100 h后(▶)、LaCo0.6Ga0.4O3(▼)和LaCo0.5Ga0.5O3(◆)样品的N2吸附-脱附等温线(a)和BJH孔径分布曲线(b)
Figure 4 N2 adsorption-desorption isotherms (a) and BJH pore size distribution (b) of the reduced LaCoO3 (■), LaCo0.8Ga0.2O3 (●), LaCo0.7Ga0.3O3 (▲) and LaCo0.7Ga0.3O3 after reaction for 100 h(▶), LaCo0.6Ga0.4O3 (▼) and LaCo0.5Ga0.5O3 (◆) samples

图 5 还原后的LaCo1-xGaxO3样品的CO2-TPD谱图
Figure 5 CO2-TPD profiles of the reduced LaCo1-xGaxO3 samples
a: x=0; b: x=0.2, c: x=0.3, d: x=0.4; e: x=0.5

图 6 反应后的LaCoO3(a),LaCo0.7Ga0.3O3(b)和还原后LaCo0.7Ga0.3O3(b′)样品的XPS谱图
Figure 6 XPS spectra of the used LaCoO3 (a), LaCo0.7Ga0.303 (b) and reduced LaCo0.7Ga0.303 (b′) samples

图 7 还原后((a)和(a′))以及反应100 h后((b)、(b′)和(b″))的LaCo0.7Ga0.303样品的TEM照片
Figure 7 TEM images of LaCo0.7Ga0.303 samples after reduction ((a) and (a′)) and after 100 h reaction test ((b), (b′) and (b″))

图 8 LaCo0.7Ga0.3O3样品反应48 h(a)、100 h(b)和LaCoO3样品反应48 h(c)的TG曲线
Figure 8 TG curves of the spent LaCo0.7Ga0.3O3 catalysts after reaction for 48 h (a) and 100 h (b) and the used LaCoO3 catalyst after reaction for 48 h (c)

图 9 还原后的LaCoO3(■)、LaCo0.8Ga0.2O3(●)、LaCo0.7Ga0.3O3(▲)、LaCo0.6Ga0.4O3(▼)、LaCo0.5Ga0.5O3(◆)样品的CO2转化率(a)、总醇选择性(b)和乙醇分布(c)随反应温度的变化
Figure 9 CO2 conversion (a), selectivity to alcohols (b), and ethanol distribution (c) as function of reaction temperature on the reduced LaCoO3 (■), LaCo0.8Ga0.2O3 (●), LaCo0.7Ga0.3O3 (▲), LaCo0.6Ga0.4O3 (▼) and LaCo0.5Ga0.5O3 (◆) catalysts

图 10 还原后LaCo0.7Ga0.3O3样品的液相分布随反应时间(h)的变化
Figure 10 Distribution of ROH products with the time on steam for CO2 hydrogenation over the reduced LaCo0.7Ga0.3O3 catalyst
表 1 LaCo1-xGaxO3样品(x=0、0.2、0.3、0.4和0.5)的元素组成、晶粒粒径、耗氢量以及还原度计算
Table 1. Elemental analysis, crystal sizes, hydrogen consumptions and reducibility degree of Con+ in the LaCo1-xGaxO3 samples (x=0, 0.2, 0.3, 0.4 and 0.5)
| Sample | Co loading/% | Co/Gaa | Co/Gab | Crystal size of Coc, d/nm | H2 uptake/ (mmol·g-1) |
Co dispersion/%c, e | H2 consumptions from TPR resultsf, g | Total theoretic H2consumptionsg | Reducibility degree of Con+/% | |
| α | β | |||||||||
| x=0 | 26.6 | - | - | 18.5 | 0.104 | 5.2 | 0.101 | 0.204 | 0.305 | 100 |
| x=0.2 | 20.6 | 4.00 | 3.97 | 8.8 | 0.184 | 11.5 | 0.077 | 0.162 | 0.242 | 98.8 |
| x=0.3 | 17.8 | 2.33 | 2.36 | 7.6(10.5)h | 0.188(0.137) | 13.4(9.8)h | 0.068 | 0.138 | 0.211 | 97.6 |
| x=0.4 | 15.0 | 1.50 | 1.50 | 6.9 | 0.173 | 14.4 | 0.064 | 0.122 | 0.180 | 103.3 |
| x=0.5 | 12.3 | 1.00 | 1.01 | 7.4 | 0.136 | 13.6 | 0.051 | 0.108 | 0.149 | 106.7 |
| a: Co/Ga ratio in synthesis system; b: Co/Ga ratio in sample measured from ICP; c: calculated from the results of H2-TPD; d: the crystal size for catalysts after reduction; e: assuming H/Co=1; f: experimental H2 consumptions calculated from TPR results using CuO as the reference material; g: the unit is mmol H2 per 50 mg of catalyst; h: crystal size of Co after 100 h stability test in the parentheses | ||||||||||
 下载: 导出CSV
下载: 导出CSV
表 2 还原后LaCo1-xGaxO3样品(x=0、0.2、0.3、0.4和0.5)的物理性质
Table 2. Physical properties of the reduced LaCo1-xGaxO3 samples
| Sample | Specific surface area A/(m2·g-1) | Total pore volume v/(cm3·g-1) | Average pore size d/nm |
| x=0 | 5.6 | 0.02 | 4.2 |
| x=0.2 | 9.6 | 0.04 | 27.0 |
| x=0.3 | 9.4(9.1)a | 0.02(0.03)a | 25.8(24.4)a |
| x=0.4 | 8.8 | 0.02 | 8.9 |
| x=0.5 | 6.5 | 0.02 | 6.2 |
| a: data in the parentheses are for the spent catalyst after 100 h reaction test | |||
下载: 导出CSV
表 3 通过XPS计算所得反应后的LaCo1-xGaxO3样品(x=0和0.3)表面组成
Table 3. Surface atomic ratios calculated by XPS results of the spent LaCo1-xGaxO3 samples (x=0 and 0.3) after reaction
| Sample | Atomic ratio/% | ||
| La/Σ[M]a | Co/Σ[M]a | Ga/Σ[M]a | |
| LaCoO3-used | 56.1(50.0)b | 43.9(50.0)b | - |
| LaCo0.7Ga0.3O3-used | 47.0(50.0)b | 15.5(35.0)b | 37.5(15.0)b |
| a: the atomic ratio of metal M is M/(La+Co+Ga) and the data in the parentheses are theoretical values | |||
下载: 导出CSV
表 4 LaCo1-xGaxO3(x=0、0.2、0.3、0.4和0.5)系列样品在CO2+H2的催化转化
Table 4. Reaction results of CO2 hydrogenation to alcohols over reduced LaCo1-xGaxO3 catalysts (x=0, 0.2, 0.3, 0.4 and 0.5)a
| Sample | CO2 conversion x/% | Selectivityb sC, mol/% | Alcohol distributionc w/% | ||||
| CH4 | C2+H | ROH | MeOH | EtOH | |||
| x=0 | 30.4 | 97.8 | 1.7 | 0.5 | 77.0 | 23.0 | |
| x=0.2 | 10.6 | 37.4 | 2.5 | 60.1 | 30.2 | 69.8 | |
| x=0.3 | 9.8 | 23.1 | 2.2 | 74.7 | 11.9 | 88.1 | |
| x=0.4 | 8.1 | 12.2 | 2.1 | 85.7 | 27.5 | 72.5 | |
| x=0.5 | 4.78 | 11.1 | 2.4 | 86.5 | - | - | |
|
a: reaction conditions: 240 ℃, 3 MPa, n(H2)/n(CO2) = 3.0, GSHV = 3000 mL/(gcat·h); the data were obtained after 18 h on stream; b: product selectivity was based carbon molar quantity, defined as the carbon molar quantity in a carbon-containing product divided by converted carbon moles; C2+H represents hydrocarbons exclusive of methane; c: alcohol distribution (mass ratio) is the weight fraction of each alcohol in total alcohols |
|||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们