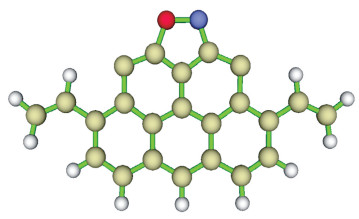
图 1.
中度气化的锯齿形碳基模型
Figure 1.
Zigzag carbonaceous model of moderate gasification char

煤炭在中国能源消费中所占比例较高[1]。燃煤电站是以煤炭作为主要的燃料,带来经济效益的同时,不可避免的带来环境污染。NOx是燃煤电站主要的一种危害性极强的大气污染物[2, 3],加之中国国家环保部发布的严格的NOx排放标准,因此,采取措施降低NOx排放,是火电厂面临的一项刻不容缓的工作。
已有的研究结果表明,氧气气氛对NO的还原有促进作用。氧对于NO还原反应的促进作用与煤焦表面的含氧官能团有关,C(O)越多,对NOx的还原程度越深[4, 5]。Suzuki等[4]实验结果表明,驻留在煤焦表面的氧显著促进了煤焦反应性。Gupta等[6]进行固定床实验,研究氧存在下的NO异相还原,结果发现氧能创造活性位点,从而促进NO还原为N2。了解污染物转化机理是控制和减少污染物排放的基础,若从反应微观角度明确煤焦异相还原NO的作用机制,就可以指导实践控制相关反应条件,最终为NOx排放控制做出实际贡献。
量子化学中的密度泛函理论日益强烈地被要求填补在均质和非均质系统的反应机制中,包括煤焦异相还原过程[7-10]。高正阳等[11]利用量子化学方法研究煤焦催化HCN还原NO的反应机理,发现了HCN还原NO的主要贡献来源于煤焦催化的异相反应。Karina等[12]对含酮煤焦表明氧的迁移、重排[13]以及对分子氧在椅型煤焦表面的化学吸附和后续反应的深入研究[14]。Zhang等[15]用密度泛函理论研究了在羟基和羰基存在下NO的非均相还原机理。
煤焦表面吸附氧原子在气化过程是必不可少的步骤,其吸附和脱附行为使煤焦表面活性位点成周期性变化,进而推动气化过程不断循环,随着气化程度的增加,煤焦表面吸附氧原子的数量增多。煤焦表面含氧基团主要以羟基、羰基、醌、羧基等形式存在于煤焦表面,其中, 羟基是煤焦中最主要的含氧基团。从已有研究来看,主要针对轻度气化煤焦(含一个羟基基团)表面NO的异相还原过程,对于中度气化煤焦对NO异相还原机理的研究报道较少。为深化对中度气化煤焦异相还原NO反应机理的研究,本研究对中度气化的七环zigzag煤焦异相还原NO的反应进行了理论计算,并对其动力学和热力学性质进行了研究,完善氧对煤焦异相还原NO反应的影响,寻求合适的反应气氛和温度条件,以达到更好的还原效果,为空气分级技术的实际应用和发展提供理论依据。
量子化学计算中单层石墨烯结构是良好的碳基模型,基于zigzag苯环簇模型的几何参数与实验所得参数相一致,因而将其作为碳基计算的理想模型[16-19]。钟俊等[20]采用七环锯齿形苯环簇结构模型研究了煤焦表面异相还原N2O反应,张秀霞[21]采用七环zigzag结构研究了焦炭对NO的异相还原。此外,大量研究表明, 焦炭表面锯齿形(zigzag)的边缘原子活性位具有较强的活性,燃烧过程中的反应大多主要在焦炭的边缘进行。因此,本研究采用改进的七元锯齿形结构模拟中度气化的煤焦表面(用含两个羟基的煤焦模型表示),研究中度气化煤焦异相还原NO的机理,进一步揭示氧对NO非均相还原的影响。具体模型结构见图 1。
量子化学中的密度泛函理论是一种广泛应用的高精度、高效率求解薛定谔方程的从头算法,已成为电子结构理论中解决许多难题的强有力的工具。已有研究表明,在B3LYP/6-31G(d)水平模拟煤焦反应,可在合理的计算成本下得到满意的结果[22]。已有的研究表明,B3LYP波函数中的自旋污染是相当小的,并且对能量性质具有极小的影响[23]。B3LYP/6-31G(d)所计算的键能和反应的热力学性质相当精确[24]。Karina等[12]采用B3LYP/6-31G(d)研究O2在石墨锯齿形表面的化学吸附。B3LYP/6-31G(d)已经成功地用于获得精确的煤焦几何结构和能量。因此,本研究选取B3LYP/6-31G(d)方法和基组组合优化得到反应物、过渡态、中间体和产物的几何构型和频率分析,并在B3LYP/6-31G(d)水平上计算单点能,在此基础上进行零点能校正。对过渡态结构的频率进行检验,过渡态有且仅有一个虚频,且虚频振动方向指向反应方向,进而进行IRC路径分析以确保反应路径正确性。本研究计算采用Gaussian09[25]软件完成。
经典过渡态理论的反应速率常数计算公式[26]如下:
|
$ {k^{{\rm{TST}}}} = \mathit{\Gamma } \times \frac{{{k_{\rm{B}}}T}}{h} \times \frac{{{Q_{{\rm{TS}}}}(T)}}{{{Q_{\rm{A}}}(T){Q_{\rm{B}}}(T)}} \times {\rm{exp}}(\frac{{-{E_a}}}{{RT}}) $ |
(1) |
式中,Γ为量子隧道修正系数;kB为Boltzmann常数,J/K;T为温度,K;h为普朗克常数,J·s;QTS、QA、QB依次为过渡态TS、反应物A和产物B的配分函数;Ea为反应势垒,kJ/mol;R为气体摩尔常数,J/(mol·K)。其中,配分函数Q是由频率计算中的振动分析得到,是平动能QT、转动能QR、振动能QV和电子能量QE的乘积,即Q=QTQRQVQE。
量子隧道修正系数Γ计算公式如下:
|
$ \mathit{\Gamma } = 1 + \left( {\frac{1}{{24}}} \right) \times {(\frac{{h{\upsilon _m}c}}{{{k_{\rm{B}}}T}})^2} $ |
(2) |
式中,υm为反应路径振动的频率,cm-1;c为光速,m/s。
然后,可以由阿伦尼乌斯公式得到在研究温度范围内的每步反应的速率常数,计算公式见式(3):
|
$ {\rm{ln}}k(T) = \frac{{-{E_{\rm{a}}}}}{R} \times \frac{1}{T} + {\rm{ln}}A $ |
(3) |
平衡常数计算公式[27]如下:
|
$ \Delta G = {G_{\rm{B}}}-{G_{\rm{A}}} =-RT{\rm{ln}}K $ |
(4) |
式中,GA和GB分别为反应物和产物的吉布斯自由能,kJ /K;K为平衡常数。
中度气化的锯齿形煤焦表面异相还原NO过程中各驻点的优化结构见图 2,反应过程中的能量变化见图 3。中度气化的zigzag形煤焦表面异相还原NO的反应,经历8个中间体和2个过渡态,最终NO被还原为N2在煤焦表面发生脱附。
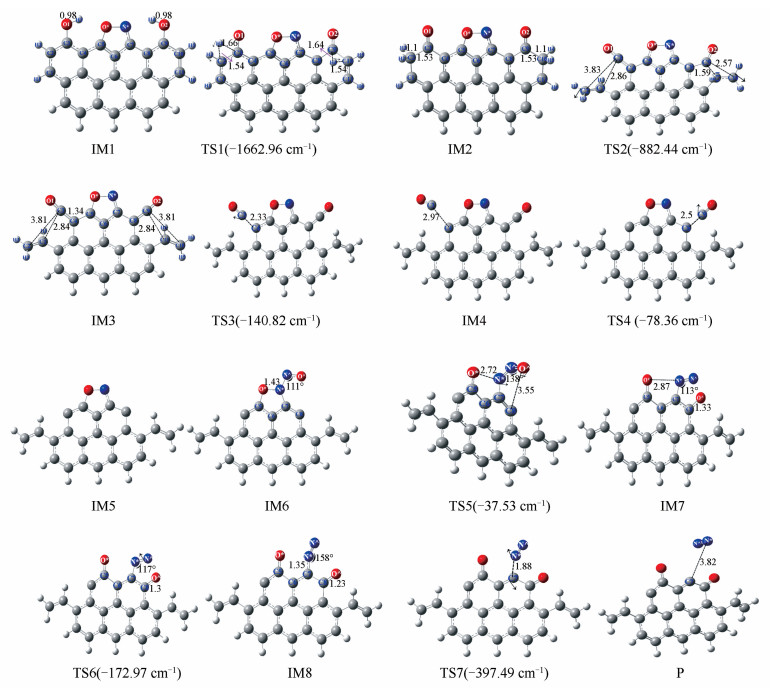
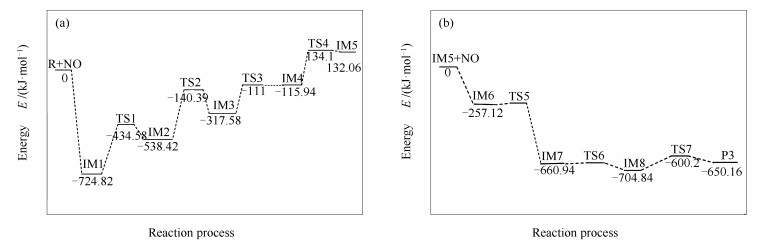
首先,含两个羟基的锯齿形煤焦以side-on的形式吸附在碳活性位表面,过程无须翻越任何能垒即可形成带有五元环结构的稳定中间体IM1,同时释放出724.82 kJ/mol的热量,吸附放出的热量可为后续反应所用。IM1翻越290.24 kJ/mol的能垒发生H原子迁移形成中间体IM2,过程中—OH对H原子作用力逐渐减弱,C2-H3和C10-H4间键逐渐增强,最终O1-H3和O2-H4键断裂,C2-H3和C10-H4键形成。IM2到IM3为六元环的开环过程,过程需克服398.03 kJ/mol的能垒,吸附过程放出的热量(Eads= -724.82 kJ/mol)可以为该步反应所用。中间体IM3较IM2能量高220.84 kJ/mol,表明IM3不稳定,将继续发生反应。IM3→IM4,CO的脱附过程,C3-C4作用力逐渐削弱,作用力的减弱利于C3-C4键的断裂。该反应的能垒为206.38 kJ/mol,需吸收201.64 kJ/mol的热量。IM4不稳定,可克服的250.02 kJ/mol能垒经TS4生成IM5,实现第二个CO在煤焦表面的脱附。由图 4中IM5的电子自旋密度图可知,IM5中C原子附近未出现自旋密度,表明电子均按自旋相反的方式配对无未成对电子。根据前线分子轨道理论,Gap的数值可以衡量分子的化学性质的稳定性,Gap越大说明分子越稳定,由表 1可知IM5的Gap值为1.254 eV,表明中间体IM5较稳定。
 下载:
导出CSV
下载:
导出CSV
| Species | HOMO/eV | LUMO/eV | Gap/eV |
| IM5 | -4.843 | -3.589 | 1.254 |
CO在煤焦表面脱附后,第二个NO以N^-down的形式吸附在N*活性位表面形成稳定中间体IM6,并放出257.12 kJ/mol的热量。IM6到IM7为O原子的迁移过程。表 2列出了IM6和IM7由NBO计算的一些重要原子的自然居群。由表 2可知,C5可以提供电子密度到受体O*,使得C5-O*键增强,并且N^可以提供电子密度到受体N*,N*-N^键增强,直接相连的N-N键为N2脱附提供基团条件。由图 3可知,IM7较IM6能量低403.82 kJ/mol,表明O原子迁移过程为放热反应,预示O^迁移基本无需克服能垒即可由N^迁移至C8活性位表面。通过N^原子绕C7-N*键旋转,IM7可经TS6转化为IM8,过程基本无需克服能垒并放出43.9 kJ/mol的热量。在图 3能量变化图中可以看出,IM7和IM8的相对能量较低,所以它们可能是稳定的中间体,在实际反应中的存在时间也可能会相对比较长。IM8克服104.64 kJ/mol的能垒将C7-N*键断裂生成产物P和N2。留在产物P结构上的O原子可进一步以CO的形式脱附、在焦炭边缘游离或参与下一步NO还原反应。
下载:
导出CSV
| Atom | Species | Charge | Valence |
| C5 | IM6 | 0.11055 | 1.87912 |
| IM7 | 0.15988 | 1.82292 | |
| O* | IM6 | -0.14716 | 3.14068 |
| IM7 | -0.31445 | 3.30572 | |
| N* | IM6 | 0.04322 | 2.44465 |
| IM7 | -0.05353 | 2.53722 | |
| N^ | IM6 | 0.12199 | 3.15905 |
| IM7 | 0.05894 | 2.42765 |
中度气化的锯齿形煤焦表面异相还原NO过程的计算结果表明:第一,NO在中度气化的锯齿形煤焦表面的吸附能(Eads= -724.82 kJ/mol)均大于其在七环锯齿形煤焦表面[21]和轻度气化的锯齿形煤焦表面吸附能[15](分别为Eads= -510.8 kJ/mol和Eads= -521.8 kJ/mol),表明中度气化的锯齿形煤焦表面更易于NO的吸附;第二,中度气化煤焦表面异相还原NO决速步为IM2→IM3,其能垒值(398.03 kJ/mol)大于七环锯齿形煤焦异相还原NO的能垒值(205.1 kJ/mol)[21],表明中度气化的煤焦不利于NO还原。中度气化的煤焦表面氧原子将与NO竞争碳基表面活性位,而由于氧的反应性更高,煤焦与氧的反应将比煤焦与NO的还原反应更有优势,从而不利于NO的异相还原。模拟计算结果与以前实验现象基本吻合,从微观分子角度验证了氧对C-NO间反应影响的实验现象。
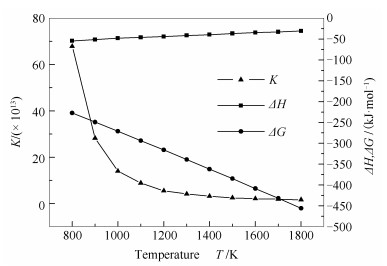
研究中度气化的锯齿形煤焦表面异相还原NO反应的热力学性质,有助于全面理解两者间化学反应。本研究选择800-1800 K计算中度气化的锯齿形煤焦表面异相还原NO的热力学参数,如焓变、吉布斯自由能的变化、反应速率常数等,这些热力学参数有利于理解反应特性,具体见图 5。
由图 5可知,在研究的温度范围内,△H随温度的升高略增大,但△H均小于0,表明中度气化的锯齿形煤焦异相还原NO反应在研究温度范围内均为放热反应。中度气化的锯齿形煤焦异相还原NO反应在研究温度范围内△G均小于0,且随温度的升高,△G逐渐减小,表明在整个煤燃烧系统中,中度气化的锯齿形煤焦异相还原NO反应均可自发进行。中度气化的锯齿形煤焦异相还原NO反应的K受温度影响较大,随温度的升高K明显降低,但在温度高于1200 K时,K基本趋于平稳,稳定在1013范围内。因此,中度气化的锯齿形煤焦异相还原NO反应在整个煤燃烧系统中,K均大于105,表明反应可以完全进行,可认为是单向反应。
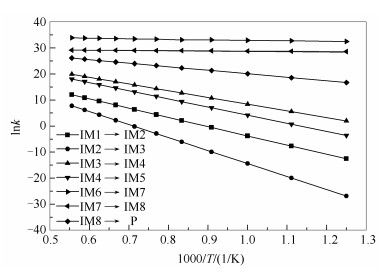
反应动力学对于评价NO还原速率具有重要意义。图 6和表 3列出了各步反应的动力学参数,图 7计算了中度气化煤焦异相还原NO总反应的速率常数。由不同温度下的反应速率常数可知,IM2→IM3的反应在相同温度下比其他反应至少慢2个数量级。因此,从动力学的观点来看,中度气化煤焦异相还原NO的速率在很大程度上取决于IM2→IM3的开环反应。由图 3中的反应过程能量变化图可知,IM2→IM3的反应所需能垒(398.03 kJ/mol)比其他反应所需翻越能垒至少高107.79 kJ/mol,则由反应过程能量变化亦可得出IM2→IM3为中度气化煤焦异相还原NO的决速步。前期研究表明,873 K时出现煤焦气化[28, 29],由图 6可知,当温度在800-900 K时开始发生反应,反应速率较小,与前期研究结论一致。当温度达到1500 K时,反应速率较大,NO的异相还原较明显。
 下载:
导出CSV
下载:
导出CSV
| Reaction | A | Ea |
| IM1→IM2 | 6.09×1013 | 294.9 |
| IM2→IM3 | 2.5×1015 | 407.4 |
| IM3→IM4 | 6.0×1014 | 212.76 |
| IM4→IM5 | 2.5×1015 | 259.8 |
| IM5+NO→IM6 | - | - |
| IM6→IM7 | 4.1×1013 | 291.17 |
| IM7→IM8 | 1.4×1015 | 408.8 |
| IM8→P | 6×1014 | 212.76 |
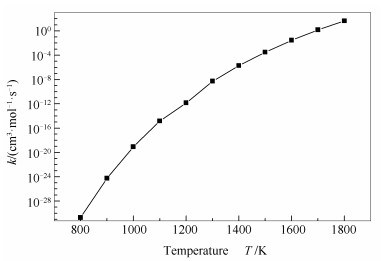
在N2形成过程中涉及的自由基表面和短暂中间体被假定为稳定状态,这种假设对于高燃烧条件下的动力学建模是合理的。由决速步理论[30]和经典过渡态理论[26]计算中度气化煤焦异相还原NO的总体反应速率常数,具体见图 7。由图 7可知,中度气化煤焦异相还原NO的阿伦尼乌斯表达式为:
中度气化煤焦异相还原NO的反应包括NO吸附、CO脱附及NO还原三个过程。结果表明,中度气化的煤焦表面更易于NO的吸附,IM2→IM3的开环过程为整个反应的决速步,所需克服能垒最大(398.03 kJ/mol)。
热力学分析表明,中度气化煤焦异相还原NO的反应在煤燃烧系统中为可自发的放热反应,且反应平衡常数大于105,表明是单向反应。
动力学分析表明,中度气化煤焦异相还原NO的阿伦尼乌斯表达式为:
付兴民, 张玉秀, 郭战英, 刘海兵, 柳树成, 贾晋炜, 舒新前. 炼焦煤尾煤热解特性及动力学研究[J]. 煤炭学报, 2013,38,(2): 320-325. FU Xing-min, ZHANG Yu-xiu, GUO Zhan-ying, LIU Hai-bing, LIU Shu-cheng, JIA Jin-wei, SHU Xin-qian. Characteristics andkinetics of the pyrolysis of coking coal tailings[J]. J China Coal Soc, 2013, 38(2): 320-325.
ZHENG M, LI X, LIU J, GUO L. Initial chemical reaction simulation of coal pyrolysis via reaxff molecular dynamics[J]. Energy Fuels, 2013, 27(6): 2942-2951. doi: 10.1021/ef400143z
苏亚欣, 苏阿龙, 成豪. 金属铁直接催化还原NO的实验研究[J]. 煤炭学报, 2013,38,(S1): 206-210. SU Ya-xin, SU A-long, CHENG Hao. Experimental study on direct catalytic reduction of NO by metallic iron[J]. J China Coal Soc, 2013, 38(S1): 206-210.
SUZUKI T, KYOTANI T, TOMITA A. Study on the carbon-nitric oxide reactionin the presence of oxygen[J]. Ind Eng Chem Res, 1994, 33: 2840-2845. doi: 10.1021/ie00035a038
YAMASHITA H, TOMITA A, YAMADA H, KYOTANI T, RADOVIC L R. Influence of char surfacechemistry on the reduction of nitric oxide with chars[J]. Energy Fuels, 1993, 7: 85-89. doi: 10.1021/ef00037a014
GUPTA H, FAN L-S. Reduction of nitric oxide from combustion flue gas bybituminous coal char in the presence of oxygen[J]. Ind Eng Chem Res, 2003, 42: 2536-2543. doi: 10.1021/ie020693n
XIN J, SUN B M, ZHU H Y, YIN S J, ZHANG Z X, ZHONG Y F. Variation analysis of Mayer bond order during the heterogeneous reduction reaction between NO and char edge models[J]. J China Coal Soc, 2014, 39(4): 771-775.
ZHOU Z, ZHANG X, ZHOU J, LIU J, CEN K. A molecular modeling study of N2 desorption from NO heterogeneous reduction on char[J]. Energy Source, 2014, 36(2): 158-166. doi: 10.1080/15567036.2010.506477
ZHU H Y, SUN B M, XI NJ, YIN S J, XIAO H P. Quantum chemistry research on NO heterogeneous reduction by char with the participation of CO under oxy-fuel combustion atmosphere[J]. J China Coal Soc, 2015, 40(7): 1641-1647.
ZHANG H, LIU J, WANG X, JIANG X. Density functional theory study on two different oxygen enhancement mechanisms during NO-char interaction[J]. Combust Flame, 2016, 169: 11-18. doi: 10.1016/j.combustflame.2016.03.023
高正阳, 杨维结, 阎维平. 煤焦催化HCN还原NO的反应机理[J]. 燃料化学学报, 2017,45,(9): 1043-1048. GAO Zheng-yang, YANG Wei-jie, YAN Wei-ping. Reaction mechanism of NO reduction with HCN catalyzed by char[J]. J Fuel Chem Technol, 2017, 45(9): 1043-1048.
KARINA S, BRIAN S H. Density functional study of the chemisorption of O2 on the zig-zag surface of graphite[J]. Combust Flame, 2005, 143(4): 629-643. doi: 10.1016/j.combustflame.2005.08.026
SENDT K, HAYNES B S. Density functional study of the reaction of carbon surface oxides:the behavior of ketones[J]. J Phys Chem A, 2005, 109(15): 3438-3447. doi: 10.1021/jp045111p
SENDT K, HAYNES B S. Density functional study of the chemisorption of O2 across two rings of the armchair surface of graphite[J]. J Phys Chem C, 2007, 111(14): 5465-5473. doi: 10.1021/jp067363r
ZHANG H, JIANG X, LIU J, SHEN J. Application of density functional theory to the nitric oxide heterogeneous reduction mechanism in the presence of hydroxyl and carbonyl groups[J]. Energy Convers Manage, 2014, 83(83): 167-176.
YANG F H, YANG R T. Ab initio molecular orbital study of adsorption of atomic hydrogen on graphite:Insight into hydrogen storage in carbon nanotubes[J]. Carbon, 2002, 40(3): 437-444. doi: 10.1016/S0008-6223(01)00199-3
CHEN N, YANG R T. Ab initio molecular orbital calculation on graphite:Selection of molecular system and model chemistry[J]. Carbon, 1998, 36(7): 1061-1070.
MIN J X, WANG N B, WANG M F, HUO P J, LIU D. Investigation on the catalytic effects of AAEM during steam gasification and the resultant char reactivity in oxygen using Shengli lignite at different forms[J]. Int J Coal Sci Technol, 2015, 2(3): 223-231. doi: 10.1007/s40789-015-0083-0
LI H B, YU Y, HAN M F, LEI Z. Simulation of coal char gasification using O2/CO2[J]. Int J Coal Sci Technol, 2014, 1(1): 81-87. doi: 10.1007/s40789-014-0010-9
钟俊, 高正阳, 丁艺, 余岳溪, 杨维结. Zigzag煤焦表面异相还原N2O反应[J]. 煤炭学报, 2017,42,(11): 3028-3034. ZHONG Jun, GAO Zheng-yang, DING Yi, YU Yue-xi, YANG Wei-jie. Heterogeneous reduction reaction of N2O by char based on Zigzag carbonaceous model[J]. J China Coal Soc, 2017, 42(11): 3028-3034.
张秀霞. 焦炭燃烧过程中氮转化机理与低NOx燃烧技术的开发[D]. 浙江: 浙江大学, 2012.ZHANG Xiu-xia. Nitrogen conversion mechanism during char combustion and develepment of low NOx technology[D]. Zhejiang: Zhejiang University, 2012.
SENDT K, HAYNES B S. Density functional study of the reaction of O2 with a single site on the zigzag edge of graphene[J]. Proc Combust Inst, 2011, 33(2): 1851-1858. doi: 10.1016/j.proci.2010.06.021
PHAM B Q, TRUONG T N. Electronic spin transitions in finite-size graphene[J]. Chem Phys Lett, 2012, 535(7): 75-79.
ALEJANDRO M, THANH-THAI T T, FANOR M, THANH N.T.. CO desorption from oxygen species on carbonaceous surface:1. Effects of the local structure of the active site and the surface coverage[J]. J Phys Chem A, 2001, 105(27): 6757-6764. doi: 10.1021/jp010572l
FRISCH M J, TRUCKS G W, SCHLEGEL H B, SCUSERIA G E, ROBB M A. Gaussian09, revision E. 01[J]. Gaussian Inc., Wallingford, CT, 2009, : .
ZHANG H, LIU J, SHEN J, JIANG X. Thermodynamic and kinetic evaluation of the reaction between NO (nitric oxide) and char(N) (char bound nitrogen) in coal combustion[J]. Energy, 2015, 82: 312-21. doi: 10.1016/j.energy.2015.01.040
ALI M A, RAJAKUMAR B. Thermodynamic and kinetic studies of hydroxyl radical reaction with bromine oxide using density functional theory[J]. Comput Theor Chem, 2011, 964: 283-290. doi: 10.1016/j.comptc.2011.01.013
PEVIDA C, ARENILLAS A, RUBIERA F, PIS J J. Synthetic coal chars for the elucidation of NO heterogeneous reduction mechanisms[J]. Fuel, 2007, 86: 41-49. doi: 10.1016/j.fuel.2006.07.002
PEVIDA C, ARENILLAS A, RUBIERA F, PIS J J. Heterogeneous reduction of nitric oxide on synthetic coal chars[J]. Fuel, 2005, 84: 2275-2279. doi: 10.1016/j.fuel.2005.06.003
GAO Z Y, LV S K, YANG W J, YANG P F, JI S, MENG X X. Quantum chemistry investigation on the reaction mechanism of the elemental mercury, chlorine, bromine and ozone system[J]. J Mol Model, 2015, 21(6): 1-9.

图 2 中度气化的锯齿形煤焦异相还原NO过程中各驻点结构
Figure 2 Geometrical structures of stationary points in the heterogeneous reduction of NO by moderate gasification char

图 3 反应过程能量的变化
Figure 3 Geometrical structures and relative energies of the stationary points

图 6 NO与中度气化煤焦反应速率常数的计算
Figure 6 Calculated thermal rate constants for the reaction between NO and moderate gasification char
表 1 IM5轨道分析
Table 1. Orbital analysis of IM5
| Species | HOMO/eV | LUMO/eV | Gap/eV |
| IM5 | -4.843 | -3.589 | 1.254 |
 下载: 导出CSV
下载: 导出CSV
表 2 由NBO计算的一些重要原子的自然居群
Table 2. Natural population of some important atoms calculated by NBO
| Atom | Species | Charge | Valence |
| C5 | IM6 | 0.11055 | 1.87912 |
| IM7 | 0.15988 | 1.82292 | |
| O* | IM6 | -0.14716 | 3.14068 |
| IM7 | -0.31445 | 3.30572 | |
| N* | IM6 | 0.04322 | 2.44465 |
| IM7 | -0.05353 | 2.53722 | |
| N^ | IM6 | 0.12199 | 3.15905 |
| IM7 | 0.05894 | 2.42765 |
下载: 导出CSV
表 3 各步反应动力学参数
Table 3. Kinetic parameters for reaction steps
| Reaction | A | Ea |
| IM1→IM2 | 6.09×1013 | 294.9 |
| IM2→IM3 | 2.5×1015 | 407.4 |
| IM3→IM4 | 6.0×1014 | 212.76 |
| IM4→IM5 | 2.5×1015 | 259.8 |
| IM5+NO→IM6 | - | - |
| IM6→IM7 | 4.1×1013 | 291.17 |
| IM7→IM8 | 1.4×1015 | 408.8 |
| IM8→P | 6×1014 | 212.76 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们