-
[1]
高爽, 陈晓陆, 汪建江, 杜国丰, 李振, 王振旅. Ru基催化剂在肉桂醛选择性加氢反应中的研究[J]. 石油化工,
2017,46,(7): 862-868.
GAO Shuang, CHEN Xiao-lu, WANG Jian-jiang, DU Guo-feng, LI Zhen, WANG Zhen-lü. Selective hydrogenation of cinnamaldehyde to cinnamyl alcohol on Ru-based catalysts[J]. Petrochem Technol,
2017, 46(7):
862-868.
-
[2]
CLAUS P, BRULCKNER A, MOHR C, HOFMEISTER H. Supported gold nanoparticles from quantum dot to mesoscopic size scale:Effect of electronic and structural properties on catalytic hydrogenation of conjugated functional groups[J]. J Am Chem Soc,
2000, 122(46):
11430-11439.
doi: 10.1021/ja0012974
-
[3]
BUS E, PRINS R, BOKHOVEN J A V. Origin of the cluster-size effect in the hydrogenation of cinnamaldehyde over supported Au catalysts[J]. Catal Commun,
2007, 8(9):
1397-1402.
doi: 10.1016/j.catcom.2006.11.040
-
[4]
HISCHL R, DELBECQ F, SAUTET P, HAFNER J. Adsorption of unsaturated aldehydes on the (111) surface of a Pt-Fe alloy catalyst from first principles[J]. J Catal,
2003, 217(2):
354-366.
doi: 10.1016/S0021-9517(03)00057-5
-
[5]
李辉, 马春景, 李和兴. Ni-Co-B非晶态合金催化肉桂醛常压加氢制3-苯丙醛的研究[J]. 化学学报,
2006,64,(19): 1947-1953.
doi: 10.3321/j.issn:0567-7351.2006.19.002LI Hui, MA Chun-jing, LI He-xing. Study on cinnamaldehyde hydrogenation to 3-phenylpropyl aldehyde at atmospheric pressure over Ni-Co-B amorphous alloys[J]. Acta Chim Sin,
2006, 64(19):
1947-1953.
doi: 10.3321/j.issn:0567-7351.2006.19.002
-
[6]
Ni X J, ZHANG B S, LI C, PANG M, SU D S, WILLIAMS C T, LIANG C H. Microwave-assisted green synthesis of uniform Ru nanoparticles supported on non-functional carbon nanotubes for cinnamaldehyde hydrogenation[J]. Catal Commun,
2012, 24(24):
65-69.
-
[7]
MARCHI A J, GORDO D A, TRASARTI A F, APESTEGUIA C R. Liquid phase hydrogenation of cinnamaldehyde on Cu-based catalysts[J]. Appl Catal A:Gen,
2003, 249(1):
53-67.
doi: 10.1016/S0926-860X(03)00199-6
-
[8]
JOSEPH ANTONY RAJ A, PRAKASH M G, ELANGOVAN T, VISWANATHAN B. Selective hydrogenation of cinnamaldehyde over cobalt supported on alumina, silica and titania[J]. Catal Lett,
2012, 142(1):
87-94.
doi: 10.1007/s10562-011-0693-0
-
[9]
LUO Q Q, WANG T, BELLER R, JIAO H J. Acrolein hydrogenation on Ni(111)[J]. J Phys Chem C,
2013, 117(24):
12715-12724.
doi: 10.1021/jp403972b
-
[10]
钱梦丹, 薛继龙, 夏盛杰, 倪哲明, 蒋军辉, 曹勇勇. Pd/Cu(111)双金属表面催化糠醛脱碳及加氢的反应机理[J]. 燃料化学学报,
2017,45,(1): 34-42.
QIAN Meng-dan, XUE Ji-long, XIA Sheng-jie, NI Zhe-ming, JIANG Jun-hui, CAO Yong-yong. Decarbonylation and hydrogenation reaction of furfural on Pd/Cu(111) surface[J]. J Fuel Chem Technol,
2017, 45(1):
34-42.
-
[11]
YUAN E, WANG L, ZHANG X W, FENG R, WU C, LI G Z. Density functional theory analysis of anthraquinone derivative hydrogenation over palladium catalyst[J]. Chem Phys Chem,
2016, 17(23):
3974-3984.
doi: 10.1002/cphc.201600874
-
[12]
ROJAS H, DIAZ G, MARTINE J J, CASTANEDA C, GOMEZ-CORTES A, ARENAS-ALATORRE J. Hydrogenation of α, β-unsaturated carbonyl compounds over Au and Ir supported on SiO2[J]. J Mol Catal A:Chem,
2012, 363(364):
122-128.
-
[13]
IDE M S, HAO B, NEUROCK M, DAVIS R J. Mechanistic insights on the hydrogenation of α, β-unsaturated ketones and aldehydes to unsaturated alcohols over metal catalysts[J]. ACS Catal,
2012, 2(4):
671-683.
doi: 10.1021/cs200567z
-
[14]
ZANELLA R, LOUIS C, GIORGIO S, TOUROUDE R. Crotonaldehyde hydrogenation by gold supported on TiO2:Structure sensitivity and mechanism[J]. J Catal,
2004, 223(2):
328-339.
doi: 10.1016/j.jcat.2004.01.033
-
[15]
LOFFREDA D, DELLECQ F, VIGNE F, SAUTET P. Catalytic hydrogenation of unsaturated aldehydes on Pt(111):Understanding the selectivity from first-principles calculations[J]. Angew Chem Int Ed,
2005, 44(33):
5279-5282.
doi: 10.1002/(ISSN)1521-3773
-
[16]
HAN Q, LIU Y F, WANG D, YUAN F L, NIU X Y, ZHU Y J. Effect of carbon nanosheets with different graphitization degree as support of noble metal on selective hydrogenation of cinnamaldehyde[J]. Rsc Adv,
2016, 6(100):
98356-98364.
doi: 10.1039/C6RA17979G
-
[17]
ABID M, PAUL-BONCOR V, TOUROUDE R. Pt/CeO2 catalysts in crotonaldehyde hydrogenation:Selectivity, metal particle size and SMSI states[J]. Appl Catal A:Gen,
2006, 297(1):
48-59.
doi: 10.1016/j.apcata.2005.08.048
-
[18]
MAHATA N, GONCALVE F, PEREIRA M F R, FIGUEIREDO J L. Selective hydrogenation of cinnamaldehyde to cinnamyl alcohol over mesoporous carbon supported Fe and Zn promoted Pt catalyst[J]. Appl Catal A:Gen,
2008, 339(2):
159-168.
doi: 10.1016/j.apcata.2008.01.023
-
[19]
TEDDY J, FALQUI A, CORRIAS A, CARTA D, LECANTE P, GERBER I, SERP P. Influence of particles alloying on the performances of Pt-Ru/CNT catalysts for selective hydrogenation[J]. J Catal,
2011, 278(1):
59-70.
doi: 10.1016/j.jcat.2010.11.016
-
[20]
JOB N, PIRARD R, MARIEN J, PIRARD J P. Porous carbon xerogels with texture tailored by pH control during sol-gel process[J]. Carbon,
2004, 42(3):
619-628.
doi: 10.1016/j.carbon.2003.12.072
-
[21]
DURNDELL L J, PARLETT C M, HONDOW N S, ISAACS M A, WILSON K, LEE A F. Selectivity control in Pt-catalyzed cinnamaldehyde hydrogenation[J]. Sci Rep,
2015, 5:
9425.
doi: 10.1038/srep09425
-
[22]
TUOKKO S, PIHKO P M, HONKALA K. First principles calculations for hydrogenation of acrolein on Pd and Pt:Chemoselectivity depends on steric effects on the surface[J]. Angew Chem Int Ed,
2016, 55(5):
1670-1674.
doi: 10.1002/anie.201507631
-
[23]
CAO X M, BURCH R, HARDCRE C, HU P. Reaction mechanisms of crotonaldehyde hydrogenation on Pt(111):Density functional theory and microkinetic modeling[J]. J Phys Chem C,
2011, 115(40):
19819-19827.
doi: 10.1021/jp206520w
-
[24]
YANG X, WANG A, WANG X, ZHANG T, HAN K, LI J. Combined experimental and theoretical investigation on the selectivities of Ag, Au, and Pt catalysts for hydrogenation of crotonaldehyde[J]. J Phys Chem C,
2009, 113(49):
97-102.
-
[25]
LI L C, WANG W, WANG X L, ZHSNG L. Investigating the mechanism of the selective hydrogenation reaction of cinnamaldehyde catalyzed by Ptn clusters[J]. J Mol Model,
2016, 22(8):
1-11.
-
[26]
JIANG Z, QIN P, FANG T. Theoretical study of NH3 decomposition on Pd-Cu(111) and Cu-Pd (111) surfaces:A comparison with clean Pd(111) and Cu(111)[J]. Appl Surf Sci,
2016, 371:
337-342.
doi: 10.1016/j.apsusc.2016.02.231
-
[27]
GOVIND N, PETERSEN M, FITZGERAID G, KING-SMITH D, ANDZELM J. A generalized synchronous transit method for transition state location[J]. Comput Mater Sci,
2003, 28(2):
250-258.
-
[28]
肖雪春, 施炜, 倪哲明. Au(111)面上肉桂醛的选择性加氢机理[J]. 物理化学学报,
2014,30,(8): 1456-1464.
doi: 10.3866/PKU.WHXB201406091XIAO Xue-chun, SHI Wi, NI Zhe-ming. Selective hydrogenation mechanism of cinnamaldehyde on Au(111) surface[J]. Acta Phys-Chim Sin,
2014, 30(8):
1456-1464.
doi: 10.3866/PKU.WHXB201406091
-
[29]
蒋军辉, 钱梦丹, 薛继龙, 夏盛杰, 倪哲明, 邵蒙蒙. In-Au(111)和Ir-Au(111)合金表面的性质及其对巴豆醛的吸附比较[J]. 物理化学学报,
2016,32,(12): 2932-2940.
doi: 10.3866/PKU.WHXB201609302JIANG Jun-hui, QIAN Meng-dan, XUE Ji-long, XIA Sheng-jie, NI Zhe-ming, SHAO Meng-meng. Comparison of properties of In-Au(111) and Ir-Au(111) alloy surfaces, and their adsorption to crotonaldehyde[J]. Acta Phys-Chim Sin,
2016, 32(12):
2932-2940.
doi: 10.3866/PKU.WHXB201609302
-
[30]
YANG X F, WANG A Q, WANG X D, ZHANG T, HAN K L, LI J. Combined experimental and theoretical investigation on the selectivities of Ag, Au, and Pt catalysts for hydrogenation of crotonaldehyde[J]. J Phys Chem C,
2009, 113(49):
20918-20926.
doi: 10.1021/jp905687g
-
[31]
PIRILLO S, LOPEZ-CORRAL I, GERMAN E, JUAN A. Density functional study of acrolein adsorption on Pt (111)[J]. Vacuum,
2014, 99(1):
259-264.
-
[32]
LIU R Q. Adsorption and dissociation of H2O on Au(111) surface:A DFT study[J]. Comput Theor Chem,
2013, 1019(1):
141-145.
-
[33]
WEI S P, ZHAO Y T, FAN G L, YANG L, LI F. Structure-dependent selective hydrogenation of cinnamaldehyde over high-surface-area CeO2-ZrO2 composites supported Pt nanoparticles[J]. Chem Eng J,
2017, 322(15):
234-245.
-
[34]
LIU H, MEI Q, LI S, YANG Y, WANG Y, LIU H, ZHENG L, AN P, ZHANG J, HAN B. Selective hydrogenation of unsaturated aldehydes over Pt nanoparticles promoted by the cooperation of steric and electronic effects[J]. Chem Commun,
2018, 54(8):
908-911.
doi: 10.1039/C7CC08942B
-
[35]
YANG B, CHERKASOV N, HUBAND S, WALKER D, WALTON R I, REBROV E. Highly selective continuous flow hydrogenation of cinnamaldehyde to cinnamyl alcohol in a Pt/SiO2 coated tube reactor[J]. Catalysts,
2018, 8(2):
58.
doi: 10.3390/catal8020058

 下载:
下载:
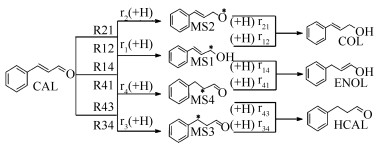
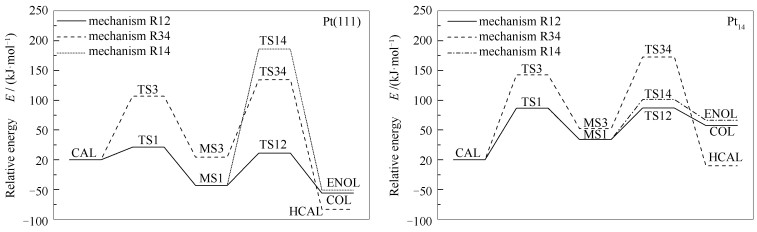

 下载:
下载:







