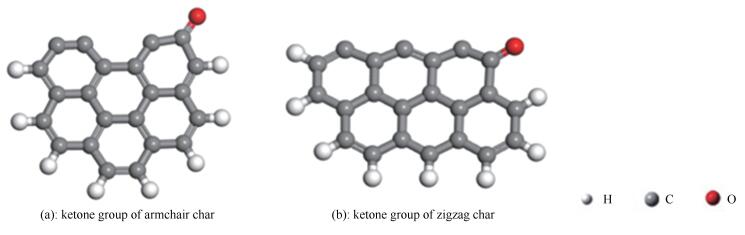
图 1.
煤焦表面模型示意图
Figure 1.
Surface of coal char

随着人们环保意识的增强及国家污染物排放标准的严格,控制燃煤过程中NOx排放已刻不容缓。已有研究表明,高温下煤焦异相还原NO是最主要的NO还原机理,温度越高,焦炭的还原作用越强。肖韡等[1]在高温条件下(温度>1300 ℃),研究了氧气对C-NO反应的影响。结果表明,氧气对C-NO反应的影响存在临界值,在临界值以下,随着O2浓度提高,煤焦对NO脱除率增大;在O2浓度为临界值时NO脱除率达到最大;此后随O2浓度升高,NO脱除率逐渐减小。
And等[2]在烟煤焦还原NO的反应系统中添加少量O2,研究结果表明,少量的O2可以增强NO的还原性能,但过高的O2则会导致煤焦表面碳活性位减少而降低脱硝率。Dong等[3]研究表明,较少的O2(0.5%)浓度仍能明显降低系统出口处NO浓度。煤焦与NO的反应依赖于煤焦表面含氧官能团,氧气主要以羟基、含酮基团、羰基等含氧官能团存在于煤焦表面,其中,含酮基团在煤焦中的含量较大,因此,研究含酮基团对煤焦异相还原NO的影响较为重要。
目前,对于氧对煤焦异相还原NO影响的研究主要依靠实验手段,对于微观机理的理论研究报道较少。近年来,量子化学在煤燃烧过程中氮元素迁移转化机理方面的研究得到了越来越多的应用。张秀霞[4]利用密度泛函理论研究NO在煤焦边缘模型表面发生异相还原的两个主要反应机理。高正阳等[5]研究了煤焦催化HCN还原NO的反应机理,研究表明,HCN还原NO的主要贡献来源于煤焦催化的异相反应。在以往的研究中采用量子化学研究了椅形和锯齿形纯碳基表面异相还原NO的过程,但含酮煤焦异相还原NO的反应路径和过渡态结构仍不清楚。本研究采用量子化学研究手段研究含酮基团对煤焦异相还原NO的影响,在分子层面计算并比较NO在椅形含酮煤焦表面与锯齿形含酮煤焦表面的吸附过程以及异相还原过程,通过研究含酮煤焦异相还原NO的反应路径,明确反应过渡态(TS)和中间体(IM)结构,揭示含酮基团对煤焦异相还原NO的影响机理。
选取煤焦-NO反应的模型结构主要集中在边缘攻击反应[6],已有研究表明,armchair构型和zigzag构型是碳质表面的两种典型研究模型[7]。文献[7, 8]用量子化学研究了石墨烯的化学性质,计算结果表明,基于六元椅形和锯齿形苯环结构的参数和实验数据吻合较好,因而更适合于反应机理研究。朱恒毅[9]研究了六元苯环椅形和锯齿形纯碳基表面异相还原NO的过程。此外,利用这两种煤焦边缘模型,成功地研究了含碳表面的许多反应机理[10-12]。因此,选择六元椅形和锯齿形苯环作为煤焦模型是合理的。本研究采用改进的椅形和锯齿形结构分别计算含酮基团对煤焦异相还原NO的影响。具体模型结构见图 1,模型中最上面一层碳原子未用氢原子封闭,用来模拟活性较好的煤焦表面,其他位置用氢原子封闭。
理论计算方法和基组的选择对计算结果的准确性有重要影响。密度泛函算法不仅计算准确,而且能解释燃烧过程中的热化学反应机理,为实际应用提供了理论支持[13, 14]。Materials Studio软件中的DMol3[15]模块拥有独特的密度泛函量子力学程序,其在模拟团簇模型上具有速率快、精度高的优势。本研究计算过程采用DMol3程序包,电子的交换关联作用选取广义梯度近似(GGA)来进行处理,所有反应物、中间体(IM)、过渡态(TS)及产物构型的优化均采用PBE泛函算法和DNP基组;自洽场(SCF)的总能量收敛极限为1.0×10-6 Ha;所有原子采用全电子计算,计算时考虑其自旋非限制性;采用“TS search”任务中的“Complete LST/QST”进行过渡态搜索,能量计算中考虑了零点能矫正;进而进行频率振动分析,通过判断振动趋势是否与反应趋势是否一致,判断所得过渡态的合理性,并保证过渡态有且仅有一个虚频。吸附能的计算公式为Eads =EA-B-(EA+EB),其中, EA-B为气体吸附后的总能量,EA为气体能量,EB为煤焦表面能量。
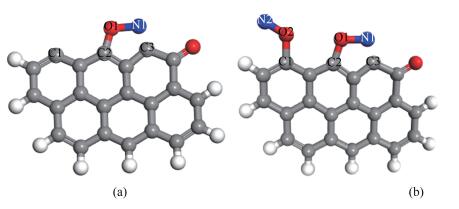
第一个NO分子以N1-down-C3的形式稳定吸附在含酮煤焦表面,吸附能最大,放出-376.21 kJ/mol热量,第一个NO分子稳定吸附后,第二个NO分子以side-on的形式平行吸附在C1、C2的活性位点,吸附结构较稳定,放出-606 kJ/mol的热量,吸附放出的高热量可为后续反应所用。朱恒毅[9]研究NO在椅形纯碳基表面吸附,放出热量为-440.28 kJ/mol的热量,本研究计算可得NO在椅形含酮煤焦表面吸附能为-982.21 kJ/mol,表明含酮煤焦表面更易于NO的吸附。这可能是因为含酮基团使得焦炭边缘活性位的电子重新排布,含酮煤焦表面的碳活性比纯碳基表面的碳活性更强。NO在煤焦表面的吸附结构见图 2。
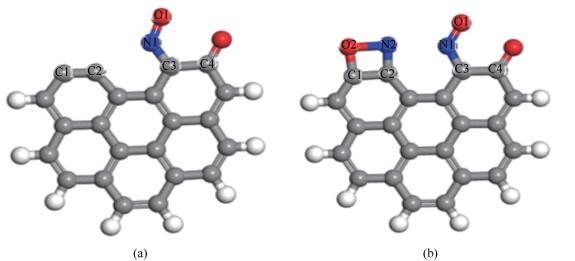
(a): N1-down-C3-CS(Eads=-376.21 kJ/mol); (b): N2O2-side-on-CS(Eads=-606 kJ/mol)
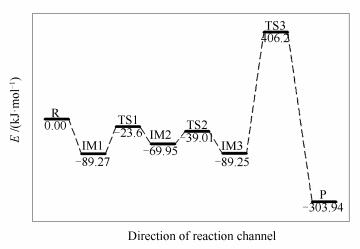
椅形含酮煤焦表面与NO反应过程和过程能量变化分别见图 3、图 4。两个NO分子稳定吸附在含酮煤焦表面,经四个过渡态,最终NO被还原为N2, 含酮基团与NO分子中的O结合生成CO2而脱离煤焦表面。整个反应过程为:首先,两个NO分子吸附在含酮armchair型煤焦表面形成稳定中间体IM1,在IM1到IM2反应过程中,O2-N2逐渐远离而发生断裂,O2-N2逐渐远离的同时伴随着N1-N2的逐渐靠近,最终与煤焦表面结合形成六元环。与此同时,含酮基团从煤焦表面脱附产生CO,经开环的过渡态TS1重组转化为更为稳定的中间体IM2。IM1到IM2过程无需能垒消耗,并且放出-86.8 kJ/mol的热量。随后IM2到IM3反应过程中,含酮基团脱附产生的CO与NO分子中的O结合,被氧化为CO2而脱离煤焦表面,过程所需克服能垒为210.9 kJ/mol,且释放出-211.92 kJ/mol热量。基于热力学数据可知,CO2从煤焦表面脱离后生成中间体IM3的结构更为稳定。IM3到IM4反应过程中,N2与C2逐渐远离而发生断裂,由六元环经过渡态TS3形成开环中间体IM4,过程需翻越410.64 kJ/mol能垒,过程基本无焓变。在图 4的反应势能面中可以看出,IM3和IM4的相对能量较低,因而它们可能是稳定的中间体,在实际反应中的存在时间可能会相对比较长。最后,IM4到产物P反应过程中,N1-C3逐渐远离而断裂,生成N2并逐渐远离煤焦表面,与此同时,C2原子与O2原子互相吸引,在该过程中最终生成一个碳氧三元杂环结构P。IM4到P过程中键长的变化为:N1-C3键长由0.1352 nm(IM4)→0.2876 nm(TS4)→0.325 nm(P)逐渐远离而发生断裂,最终生成N2而脱离煤焦表面;C2-O2键长由0.2412 nm(IM4)→0.1991 nm(TS4)→0.1436 nm(P)逐渐靠近而结合为碳氧三元杂环结构(O2-C1-C2)。过程所需克服能垒为439.41 kJ/mol,NO吸附所放出的热量(Eads=-982.21 kJ/mol)及上述前两步反应所放出热量(ΔH1=-86.8 kJ/mol,ΔH2=-211.92 kJ/mol),可以为该步所需高能垒所用。过程焓变为389.61 kJ/mol,可知该步反应为吸热反应。留在产物P结构上的O原子可进一步以CO的形式脱附或在焦炭边缘游离。
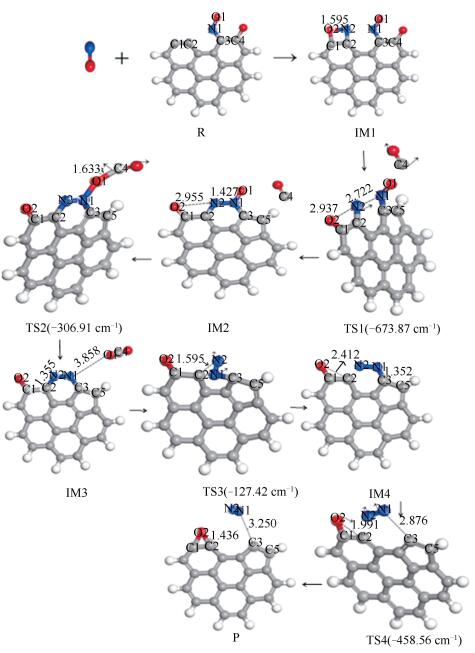
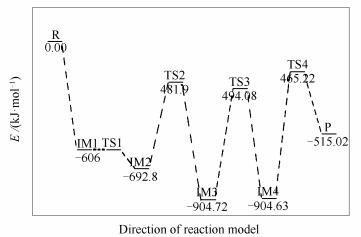
椅形含酮煤焦与椅形纯碳基煤焦异相还原NO的过程[9]的计算结果表明,椅形含酮煤焦表面更易于NO的吸附(Eads=-982.21 kJ/mol),这可能是因为含酮基团使得焦炭边缘活性位的电子重新排布,含酮煤焦表面的碳活性比纯碳基表面的碳活性更强。各反应路径中的最高能垒步即为整个反应中的决速步,则椅形含酮煤焦异相还原NO的决速步能垒值为439.41 kJ/mol,小于椅形纯碳基煤焦异相还原NO的决速步能垒值(478.1 kJ/mol),表明椅形含酮基团强化了煤焦异相还原NO。部分试验研究也证实氧的存在会在一定程度上加快在焦炭表面的还原[2, 3]。
锯齿形煤焦与NO的反应可分为两个阶段,NO分解阶段和剩余氧脱附阶段。具体地说,剩余氧脱附阶段取决于是否存在CO。该非均相还原的第一步是NO在煤焦边缘的吸附过程,气体浓度对煤焦的竞争吸附过程有重要影响。然而,探究煤焦与吸附在其边缘的气体间的反应机理是本研究的主要目的。因此,本研究不考虑气体浓度对煤焦异相还原NO的影响。
第一个NO分子以O1-down-C2形式吸附在含酮煤焦表面时,吸附能最大,放出-663.05 kJ/mol能量。第一个NO分子稳定吸附后,第二个NO分子以O2-down-C1的形式吸附在煤焦表面,放出-89.27 kJ/mol热量。吸附放出的高热量可为后续反应所用。张秀霞[4]研究NO在锯齿形纯碳基表面吸附,放出热量为-510.8 kJ/mol的热量,本研究计算可得NO在锯齿形含酮煤焦表面吸附能为-752.32 kJ/mol,表明含酮煤焦表面更易于NO的吸附。NO分子在锯齿形含酮煤焦表面稳定的吸附形式见图 5。
(a): O1-down-CS(Eads=-663.05 kJ/mol); (b): O2-down-CS(Eads=-89.27 kJ/mol)
含酮煤焦表面与NO反应过程和过程能量变化分别见图 6、图 7。两个NO分子均以O-down的形式吸附在煤焦表面,经三个过渡态,最终NO被还原为N2脱离煤焦表面。整个反应过程为:首先,两个NO分子均以O-down形式吸附在zigzag形含酮煤焦表面形成稳定中间体IM1,在IM1到IM2反应过程中,O1-N1逐渐远离而发生断裂,O1-N1键长由0.1435 nm(IM1)→0.210 nm(TS1)→0.2688 nm(IM2),反应过程中所需克服能垒为65.67 kJ/mol,反应过程焓变为19.32 kJ/mol;随后IM2到IM3反应过程中,O2-N2逐渐远离而发生断裂,O2-N2键长由0.1401 nm(IM2)→0.2804 nm(TS2)→0.2903 nm(IM3),过程所需克服能垒为30.94 kJ/mol,反应焓变为-19.3 kJ/mol,中间体IM3较IM2稳定;最后,IM3到产物P反应过程中,N1-N2两原子逐渐靠近生成N2,生成的N2并逐渐远离煤焦表面,该过程所需克服能垒为495.45 kJ/mol,NO吸附所放出的高热量(Eads=-752.32 kJ/mol)可以为该步所需高能垒所用,过程焓变为-214.69 kJ/mol,可知该步反应为放热反应。在图 7的反应势能面中可以看出,P1相对能量较低,因而可能是稳定的中间产物,在实际反应中的存在时间可能会相对比较长。
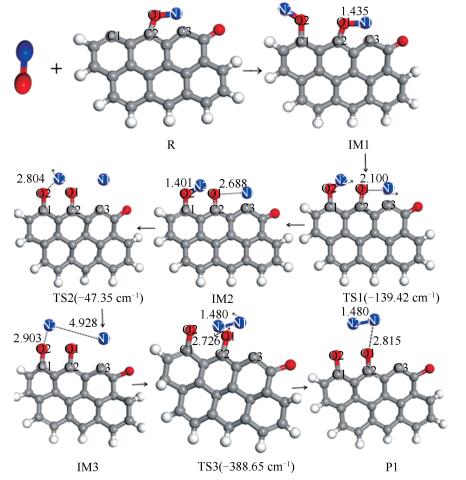
锯齿形含酮煤焦与NO还原的计算结果表明:NO分子在锯齿形含酮煤焦表面的吸附能(Eads=-752.32 kJ/mol)明显大于NO分子在锯齿形纯碳基煤焦表面的吸附能(Eads=-510.8 kJ/mol)[4],表明含氧煤焦吸附能更大,结构更稳定,更易于NO的吸附。这可能是因为氧原子使焦炭边缘活性位的电子重新排布,含酮碳表面的碳活性位比纯碳基表面的碳原子活性更强。锯齿形含酮煤焦与NO异相反应的决速步是反应的第三阶段,过程反应能垒值(495.45 kJ/mol)大于锯齿形纯碳基煤焦表面与NO反应决速步能垒数值331.32 kJ/mol[9],计算结果表明,锯齿形含酮煤焦表面不易于NO的还原。原因可能是因为含酮煤焦模型中的氧浓度不在利于NO还原的范围内,从而导致N2脱离煤焦表面需翻越更高的能垒。
在含酮煤焦表面与NO异相反应的基础上,研究了其产物表面氧的脱附机理。椅形含酮煤焦异相还原NO的产物P中的O原子,生成边缘具有三元环结构(C1-O2-C2)的中间产物P,研究表明该中间体结构稳定,留在产物P结构上的O原子发生CO的脱附需克服较高能垒[4]。在CO存在条件下,P中三元环结构C1、O2和C2均达到饱和,CO只能与C3剩余活性位点结合,并不会继续发生氧脱附过程。本研究针对P1(锯齿形含酮煤焦异相还原NO的产物)发生表面缺陷以及CO存在条件下氧脱附的微观反应机理进行研究,分别研究其发生表面缺陷以及CO存在条件下氧脱附的微观反应机理。基本反应可以表述如下:
|
$ {\rm{C}}\left( {\rm{O}} \right) \to {\rm{Cd }} + {\rm{ CO}} $ |
(1) |
|
$ {\rm{CO + C}}\left( {\rm{O}} \right) \to {\rm{C}}{{\rm{O}}_{\rm{2}}}{\rm{ + C()}} $ |
(2) |
其中,C(O)表示含氧煤焦,Cd表示煤焦边缘缺陷,C()表示含自由活性位点的煤焦表面。
文献[16, 17]用量子化学计算研究了石墨烯模型的化学性质,并表明由于锯齿形煤焦模型的优良参数与实验数据吻合,因而更适合于反应机理的研究。此外,锯齿形煤焦表面模型已经成功应用到许多碳表面反应机理的研究[18-20]。但用量子化学研究含官能团的煤焦表面发生氧脱附的机理报道较少,因此,本研究在锯齿形含酮煤焦表面与NO异相反应的基础上,对其产物P1继续研究了发生表面缺陷以及CO存在条件下氧脱附的微观反应机理。
反应物、IM、TS和产物的优化结构及反应路径见图 8,相应的反应势能见图 9。
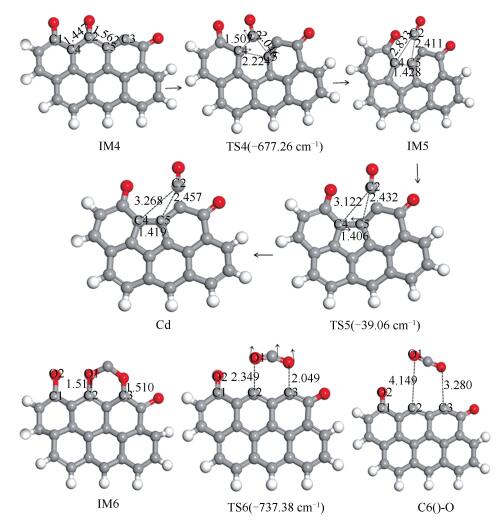
a: without CO; b: with CO
P1表面根据是否有CO的存在,有两种不同的氧脱附机理,无CO存在时的反应势能见图 9a,有CO存在时的反应势能见图 9b。
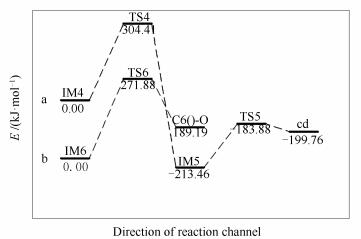
当没有CO存在的情况下,需经历两个过渡态,才能完成P1表面氧脱附过程。反应经过两个过程:首先,P1表面O1与C2结合,形成CO,过程所需能垒消耗为304.41 kJ/mol,过程焓变为-213.46 kJ/mol;随后,C3和C5原子结合,形成含酮的煤焦边缘缺陷模型Cd,过程克服能垒值为29.58 kJ/mol,过程焓变为13.7 kJ/mol。P1产生表面缺陷的具体步骤为:第一个过程中C2-C4、C2-C5逐渐远离而断裂,与此同时C4-C5逐渐靠近;第二个过程中C3和C5原子键长由0.1428 nm(IM5)逐渐靠近到0.1406 nm(TS5)最终稳定到0.1419 nm(Cd),形成含酮的煤焦边缘缺陷模型Cd,同时生成的CO逐渐远离煤焦表面。
当有CO存在情况下,过程只需经历一个过渡态,即可完成P1表面氧脱附过程。具体反应过程为:CO吸附到P1表面形成六元闭环的中间体IM6,随后闭环中的O1-C2、O-C3逐渐远离最终生成CO2而脱离P1表面,过程克服能垒为271.88 kJ/mol,过程总能量变化为189.19 kJ/mol。
前期研究结果表明,CO脱离锯齿形纯碳基煤焦表面需克服361.01 kJ/mol的能垒。事实上,Chaparala等[21]理论研究表明活化能对邻近基团有高度依赖性。Singh等[22]发现基于临近基团及游离位置不同,CO脱附的活化能在251-360 kJ/mol。P1表面氧发生脱附形成CO,过程决速步能垒消耗值为304.41 kJ/mol,计算结果与文献[21, 22]的结果一致,表明含氧煤焦易于CO的脱附而发生表面缺陷。
本研究结果可知,在无CO情况下,P1表面氧脱附过程决速步能垒消耗值为304.41 kJ/mol,高于有CO存在条件下氧脱附过程能垒值271.88 kJ/mol。结果表明,CO存在条件下更利于P1表面的氧脱附过程。
NO在椅形和锯齿形含酮煤焦表面的吸附能分别为-982.21和-752.32 kJ/mol,含酮煤焦表面更易于NO的吸附。
无CO条件下,含氧煤焦更易于发生氧脱附而产生表面缺陷;CO存在条件下,P1作为重要的催化剂,一方面, 为反应提供自由活性位点; 另一方面, 降低了反应能垒消耗,CO存在条件下更利于P1表面的氧脱附过程。
椅形含酮基团强化了煤焦异相还原NO,表明氧在一定程度上强化了煤焦异相还原NO的过程;锯齿形含酮煤焦异相还原NO需克服较高能垒(495.45 kJ/mol),表明锯齿形含酮煤焦表面不易于NO的还原。究其原因是含酮基团的armchair结构和zigzag结构活性位特性不同的原因,使得含酮椅形煤焦和含酮锯齿形煤焦结构活性位在电子排布、电荷密度及电磁特性方面均有很大差异。

Figure 2 Adsorption configurations of NO on ketone of armchair char surface
(a): N1-down-C3-CS(Eads=-376.21 kJ/mol); (b): N2O2-side-on-CS(Eads=-606 kJ/mol)

Figure 4 Potential energy of heterogeneous reduction of ketone of armchair char and NO

Figure 5 Adsorption configurations of NO on ketone of zigzag char surface
(a): O1-down-CS(Eads=-663.05 kJ/mol); (b): O2-down-CS(Eads=-89.27 kJ/mol)

Figure 8 Surface defects of P1 and the mechanism of oxygen removal with the presence of CO

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:



