 Figure 1.
Reaction of BOB and isopropanol
Figure 1.
Reaction of BOB and isopropanol
Citation:
LEI Zhao, JIANG Jing, ZHU Gang-li, ZHAO Zhi-gang, LING Qiang, CUI Ping. Investigation on the reactivity of isopropanol with lignite-related model compound[J]. Journal of Fuel Chemistry and Technology,
2016, 44(1): 7-14.

褐煤模型化合物与异丙醇进行醇解的反应性研究
English
Investigation on the reactivity of isopropanol with lignite-related model compound
Abstract:
The isopropanolysis of lignite model compound was investigated using the density functional theory method. Firstly, thermodynamic properties were estimated. Secondly, the method combined the Hirshfeld population and the Fukui function was proposed to obtain the initial reactant configuration. Thirdly, the Linear Synchronous Transit method combined with the Quadratic Synchronous Transit method was developed to calculate the reaction pathway and simultaneously optimize the structures of reactant and product. It was observed that the calculated enthalpy was decreased with increasing temperature. Furthermore, the nucleophilic group was discovered. Moreover, it was proved that the isopropanol was the most active among the common alcohols, indicating that the isopropanolysis was exothermic and nucleophilic.
-
Key words:
- isopropanolysis
- / lignite
- / optimization
- / density functional theory
- / reaction mechanism
-
Lignite as fuel is uneconomic due to high ash yield, poor stability and low calorific[1]. In fact, there is high oxygen content in lignite. Therefore, many high value-added oxygen-containing chemicals can be obtained from lignite, which caused a wide range of concern[2].
Lignite degradation in the proper solvent is an important step to obtain the high value-added oxygen-containing chemical. It was reported that methanol, ethanol and isopropanol are effective solvents because of high hydrogen donating and alkylating abilities[3-5]. Therefore, many literatures of lignite degradation with alcohols have been published. Lei et al[6]studied the degradation behavior of Shengli lignite with methanol, and revealed that Shengli lignite had a good reaction activity. Kuznetsov et al[7] investigated the reactivity of Kansk-Atchinsk lignite with two lower aliphatic alcohols, and demonstrated that those alcohols (ethanol and isopropanol) were active. Further, the mechanism of lignite degradation had been investigated. First, it was found that the more soluble portion was obtained using branched alkanol than the straight one[8]. Lu et al[9] confirmed that the nucleophilic attack of the oxygen atom in alkanol took place in the degradation of Huolinguole lignite, and proved that the nucleophilic reaction is crucial reactive step to produce the high value-added oxygen-containing chemical. In addition, it was reported that the radical formation was not involved in the alkanolysis[2]. Unfortunately, the mechanism of lignite degradation with isopropanol at the molecular level is rarely investigated.
The reaction mechanism at the molecular level was usually investigated using theoretical chemistry methods[10]. Among those methods, the density functional theory (DFT) was one of the most popular methods to explore the reaction mechanism at the molecular level[11]. However, the DFT method was completely based on a rigorous initial structure[12]. And then, the initial molecular structures were prominent during DFT calculation process. There were some ways to obtain information of the initial structures. Firstly, the number of electrons associated with the bond was obtained by using the Hirshfeld population analysis[13]. Then, the Fukui function method revealed the reactivity of a molecule with respect to electrophilic and nucleophilic attack on the charge density[14]. Combined two methods together, the relative configuration of reagents was acquired. However, the configuration using two above methods was coarse during the reaction mechanism investigation. Therefore, Linear Synchronous Transit (LST) combined with the Quadratic Synchronous Transit (QST)[15] was also proposed for optimizing reactant structure and simultaneously obtaining the transition state[16, 17].
On the other hand, the thermodynamic properties reflected equilibrium characteristic of reaction[18], which was calculated based on the vibration analysis using DFT method[19-21]. It has been reported that the reaction enthalpies (H) related to three mechanisms of phenolic antioxidants action in gas-phase were calculated using the DFT method[22]. Similarly, values of entropies (S), free energy (G) and heat capacity at constant pressure (Cp) were also obtained with the DFT method. Based on the above datum, the equilibrium constant was then estimated[23]. However, literature including thermodynamic properties analysis and reaction mechanism of lignite degradation was seldom published.
Lignites have macromolecular structures and contain amounts of oxygen-containing moieties[24]. Lignin during the coal-forming process is an important moiety and underwent less change due to its structural stability[25]. Benzyloxybenzene (BOB) is an important moiety in brown coal, and was used as lignin structure model published by Adler[26]. In addition, BOB exists in macromolecular structure of lignite, and plays the significant role in coal conversion. Therefore, BOB was chosen as lignite-related model compound to study the characteristic of lignite isopropanolysis.
As the mentioned discussion, the reactivity of BOB and isopropanol was investigated in our work. First, thermodynamic properties of BOB isopropanolysis were estimated based on the vibration analysis using the DFT method. Then, the configuration of BOB and isopropanol was determined based on Hirshfeld electron and Fukui function. And, the improved method based on the LST/QST was carried out to obtain the transition state and simultaneously optimize structures of reactant and product.
1 Methodology
As showed in Figure 1, the reaction between BOB and isopropanol is reversible. To investigate the characteristics of this reaction, the Dmol3 module of Materials Studio 6.0 package was introduced.
Figure 1.
Reaction of BOB and isopropanol
Parameters of calculation process were described as follows: Generalized gradient approximation, Becke and Perdew functional, double numerical plus polarization, and Fine and Octupole were installed as the method, functional, basis set, self-consistent field tolerance and aux-density, respectively. The initial configuration of reactants was obtained according to the Hirshfeld electron analysis combined with Fukui function. Then, the configuration of products was got based on the initial reactants. The transition state (TS) search was performed on the above theoretical level with LST and QST. The LST maximization was performed with an energy minimization method to obtain the approximated TS. After that, the approximated TS were optimized using the QST method to get the point with only one imaginary frequency. To completely study the reaction path, the intrinsic reaction coordinate (IRC) analysis was performed using the transition state (TS) confirmation tool. Then, the minimum energy path was calculated based on the IRC theory. The convergence criterion for the TS searches was set to 0.01 Hartree/Å with the root-mean-square of atomic forces. Vibrational frequencies were carried out to calculate frequencies of all initial and final states as well as the TS from the Hessian matrix. Then, the zero-point energy (ZPE) was also estimated on the basis of vibrational frequencies results.
In addition, thermodynamic properties such as G, H, S and Cp are important to understand the reaction equilibrium. What is more, the change of Gibbs free energy is pivotal to realize reaction equilibrium behavior. Therefore, the Gibbs free energy is calculated using H, S and Cp as shown in Figure 2.
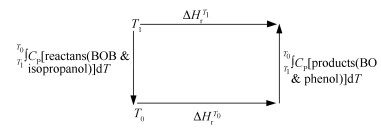 Figure 2.
Pathway of estimated Gibbs free energy T0=0 K
Figure 2.
Pathway of estimated Gibbs free energy T0=0 K
As shown in Figure 2, the enthalpy of reaction at temperature T1 was calculated using Eq. (1).
The Gibbs free energy change is given by Eq. (2).
where ΔS can be obtained from the population analysis in Materials Studio 6.0.
Then, the equilibrium constant can be estimated using Eq. (3).
2 Results and discussion
2.1 Analysis of thermodynamic properties
Thermodynamic properties of components on the above calculation level from 300 to 875 K in steps of 25 K are calculated and presented in Figure 3.
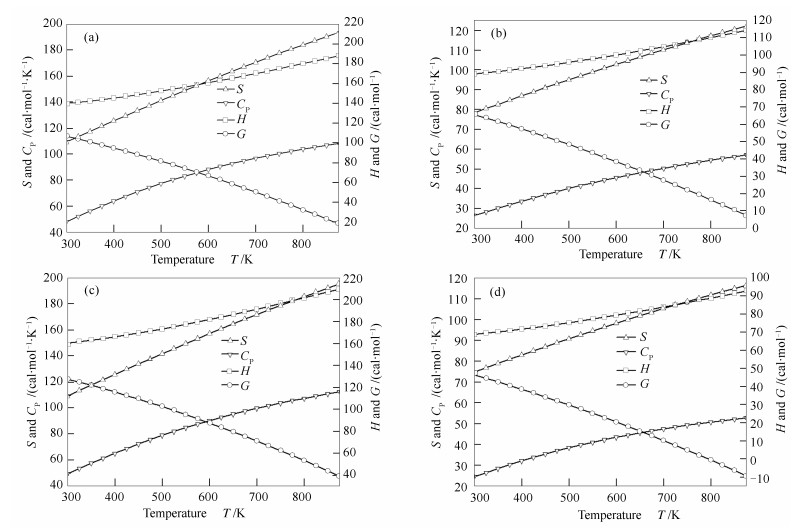 Figure 3.
Thermodynamic properties of components
Figure 3.
Thermodynamic properties of components
Based on those estimated datum, Gibbs free energy change and equilibrium constants are calculated using Eqs.(2) and (3), respectively. Then, equilibrium constant from 300 to 875 K in steps of 25 K is shown in Figure 4. The scatter point is the calculated data with Materials Studio 6.0 software. The solid line represents the estimated value expressed as Eq. (4).
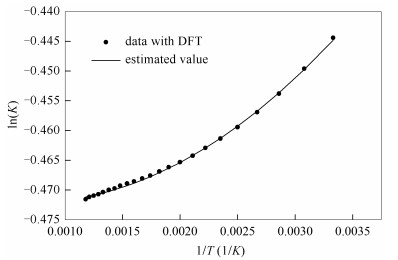 Figure 4.
Plotting for the ln K vs. 1/T
Figure 4.
Plotting for the ln K vs. 1/T
As showed in Figure 4, it is concluded that the equilibrium constant is decreased with increasing temperature, which implies that lignite isopropanolysis is exothermic. The conclusion is consistent with the reported literature[2].
2.2 Initial configuration of reactants
Since the Hirshfeld population analysis provides a reasonable representation of the electrostatic potential for molecular simulation[27], the charge definition is selected in our investigation. Based on the analysis of the density matrix, Hirshfeld charge is obtained and presented in Figure 5.
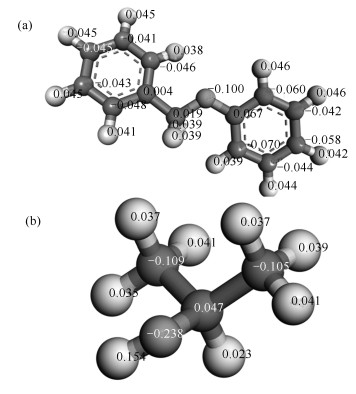 Figure 5.
Hirshfeld charge distribution of reactants
Figure 5.
Hirshfeld charge distribution of reactants
As showed in Figure 5(a), the maximal value of the negative charge is -0.100 on the oxygen atom. In addition, the maximal value of the positive charge is 0.154 on hydrogen atom as showed in Figure 5(b). Therefore, it was clearly seen that the new polar bond (H-O) is formed because the electronegativity of oxygen is greater than hydrogen.
The reactivity of chemical species is consulted on their intrinsic electronic properties. Many methods such as atomic charge computation[28], spin populations[29] and Laplacian of the charge density[30] were employed to get this property. Among those methods, Fukui function based on the frontier orbital theory was the most successful method[31]. Fukui function related the reactivity of a molecule with respect to electrophilic and nucleophilic attack on the charge density. In detailed, the sensitivity of the charge density ρ(r) was measured using the Fukui function with respect to the loss or gain of electrons via following equations[31].
where f+(r) measures changes in the density when the molecule gains electrons, which is corresponds to reactivity with respect to nucleophilic attack. On the contrary, f-(r) corresponds to reactivity with respect to electrophilic attack (loss of electrons). Moreover, radical attack f0(r) is simply the average of the two with respect to free radical attack and ΔN is the change of a number of electron.
Based on Lu's investigation[9], lignite isopropanolysis is the nucleophilic reaction. Therefore, the reactivity of reactants is estimated using the f+(r) function and represented in Figure 6.
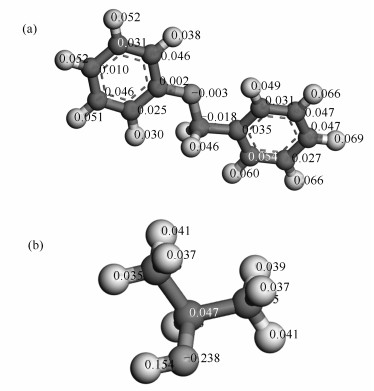 Figure 6.
Charge distribution of reactants using the Fukui function
Figure 6.
Charge distribution of reactants using the Fukui function
As shown in Figure 6, it is found that the maximal positive value in isopropanol molecule is 0.154 on the hydrogen atom connected with oxygen, and the negative charge is -0.003 on the oxygen atom in BOB molecule. Furthermore, it is inferred that oxygen in BOB molecule is close to the hydrogen atom connected with oxygen in isopropanol, and -CH2 in BOB is near to oxygen in isopropanol. Thus, the initial confirmation of reactants is illustrated in Figure 7(a). By slightly changing the position of isopropanol and BOB, the confirmation of products is shown in Figure 7(b).
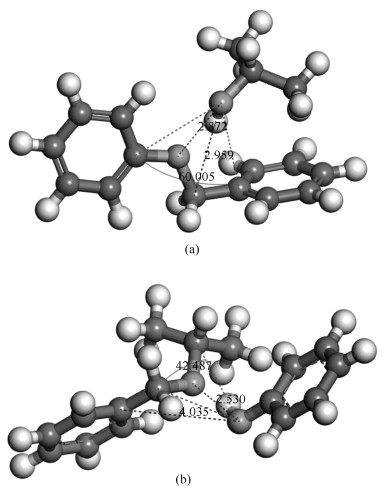 Figure 7.
Initial configuration of reactants (a) and products (b)
Figure 7.
Initial configuration of reactants (a) and products (b)
2.3 Mechanism of reaction
When the initial configuration of reactants was confirmed, the corresponding configuration of products was also determined. Then, information of reaction pathway was obtained by Reaction Preview Tool and the TS was estimated using the complete LST/QST method. Simultaneously, molecular configurations of reactants and products were optimized.
2.3.1 Transition state
The TS with only one imaginary frequency (638.45 cm-1) is obtained and showed in Figure 8. The charge of hydrogen connected with oxygen and oxygen in isopropanol molecule is 0.400 and -0.684, respectively. The charge of oxygen in BOB molecule is -0.619. The distance between hydrogen connected with oxygen in isopropanol and oxygen in BOB is 0.18 nm, which is close to a carbon single bond length (about 0.154 nm). Therefore, the hydrogen atom in isopropanol molecule is attracted with oxygen in BOB molecule to form the new O-H bond. Because there is a double bond between methylene and the positive carbon (0.075) in the benzene ring, the oxygen atom in isopropanol molecule is close to the carbon atom in methylene to form the new C-O bond. Based on structural transformation and imaginary frequency, it is proved that the transition state is reasonable.
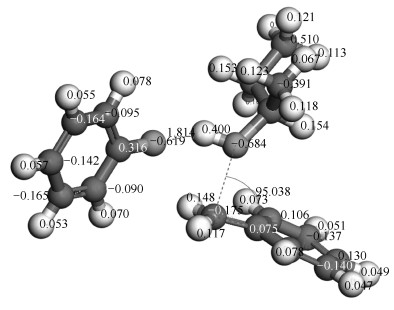 Figure 8.
Transition state
Figure 8.
Transition state
2.3.2 Pathway of reaction
According to the above TS structure, the energy profiles using the TS search tool are shown in Figure 9. Then, the schematic energy profile of lignite isopropanolysis with TS confirmation is presented in Figure 10. It is found that TS energy using TS search and TS confirmation is -771.337 25 and -771.393 45 Ha, respectively. The relative error between two values is 0.073% based on kJ/mol, indicating that the TS search is successful. As shown in Figure 10, it is concluded that the activation energy of the forward and the negative direction is 100.35 and 164.74 kJ/mol, respectively. In other words, activation energy of isopropanolysis is 100.35 kJ/mol. It was reported that the activation energy of BOB reacted with methanol and ethanol was 175.5 and 163.0 kJ/mol, respectively[2]. Therefore, it is evident that activation energy is decreased with the order methanol > ethanol > isopropanol. Thus, it is concluded that isopropanol is the most active among three alcohols, which is well agreement with the published literature[3]. These results indicate that the estimated activation energy is valid.
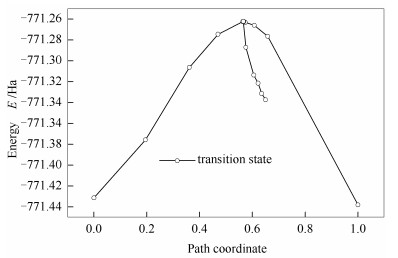 Figure 9.
Calculated energy profiles using TS search tool
Figure 9.
Calculated energy profiles using TS search tool
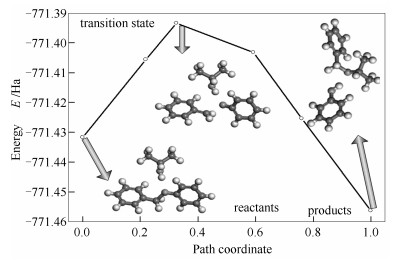 Figure 10.
Minimum energy path using TS confirmation tool
Figure 10.
Minimum energy path using TS confirmation tool
To further verify the calculated data, the rate constants of isopropanolysis at the range from 300 to 875 K with 5 K step are estimated using the following Eq. (7) and shown in Figure 11.
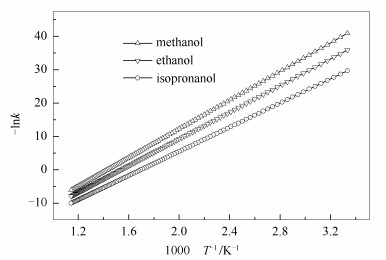 Figure 11.
Arrhenius plots for reaction of BOB and different alcohol
Figure 11.
Arrhenius plots for reaction of BOB and different alcohol
where kb, T, h, R, p0, Ea, and n denote Boltzmann constant, reaction temperature, Plank constant, universal gas constant, standard atmospheric pressure, activation energy and the mole number of reactants, respectively.
As presented in Figure 11, the k of BOB ethanolysis is higher than that of methanolsis at the same temperature. Moreover, the k of BOB reacted with isopropanol is more than that of ethanolysis at the same temperature. This result is consistent with the publication[5]. In addition, it was well known that the K was equal to the rate constant of the forward reaction dived by the reverse reaction one. Using Eq. (7), the K at 298 K is 0.690 1 and the K at the same temperature is 0.641 3 using Eq. (4), indicating that the calculated values are available.
Based on above results, the reaction mechanism is presented in Figure 12. As is evident from Figure 12, the oxygen atom in BOB is attracted with the hydrogen of hydroxy in isopropanol. And the oxygen atom in isopropanol is allured with the carbon atom of methylene in BOB. Owing to those attractive forces, two molecules are close to each other. When the transition state is reached, it has been inferred that the attractive force still appears, but less than the initial structure. Because the attractive force appears in the reactive process, the nucleophilic reaction takes place. According to the mechanism of the nucleophilic reaction, the new H-O bond is formed by oxygen atom in BOB and hydrogen of hydroxy in isopropano. Then, carbon atom of methylene in BOB and oxygen atom in isopropanol are connected to form the new C-O bond. Thus, the products are obtained on the foundation of this reaction mechanism.
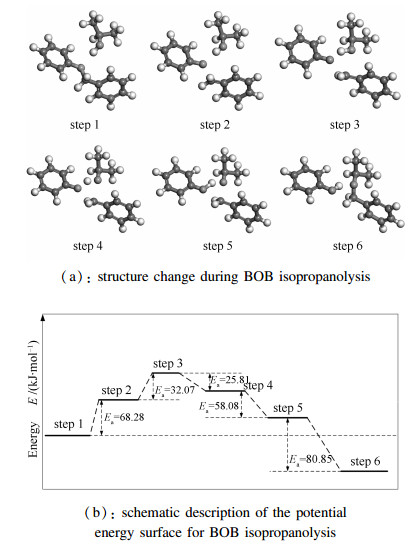 Figure 12.
Mechanism of reaction
Figure 12.
Mechanism of reaction
3 Conclusions
The reactivity of lignite isopropanolysis using benzyloxybenzene as the model compound was investigated with the DFT method. By analyzing the vibration, it was found that the lignite isopropanolysis is exothermic. Moreover, the initial structure of reactants was obtained using the Fukui function combined with Hirshfeld population analysis. Then, the LST and QST method was proposed to estimate the transition state, and simultaneously to optimize structures of reactant and product. After that, reaction pathway and activation energy were calculated with the transition state confirmation tool. By comparing the estimated activation energy with the reported one, it was concluded that the estimated activation energy was reasonable and valid. Furthermore, the nucleophilic reaction mechanism was clarified at the molecular level, which was in agreement with the literature reported[2].
Notation
Cp: heat capacity at constant pressure (cal·mol-1·K-1)
Ea: activation energy (kJ·mol-1)
f0(r): the Fukui function with free radical attack
f+(r): the Fukui function with nucleophilic attack
f-(r): the Fukui function with electrophilic attack
G: the free energy (cal·mol-1)
h: Plank constant
H: the reaction enthalpy (cal·mol-1)
k: the rate constant
kb: Boltzmann constant
K:equilibrium constant
n: the mole number of reactants
ρ(r): the charge density
p0: standard atmospheric pressure (atm)
S: the entropy (cal·mol-1·K-1)
T: reaction temperature (K)
ΔN: the change of a number of electron
-
-
[1]
CHATTERJEE K K. Uses of energy, minerals and changing techniques[M]. New Age International (P) Ltd, 2006.
-
[2]
LI K Z, ZONG M Z, YAN L H. Alkanolysis simulation of lignite-related model compounds using density functional theory[J]. Fuel, 2014, 120: 158-162. doi: 10.1016/j.fuel.2013.12.009
-
[3]
KUZNETSOV N P, SHARYPOV I V, RUBAYLO I A. Study of Kansk-Atchinsk lignite liquefaction in lower aliphatic alcohols using flow periodical function[J]. Fuel, 1988, 67(12): 1685-1690. doi: 10.1016/0016-2361(88)90217-7
-
[4]
KUZNETSOV N P, SHARYPOV I V, BEREGOVTSOVA N G. Kinetics and isotope effect of brown coal liquefaction in ethanol[J]. React Kinet Catal Lett, 1989, 40(1): 59-64. doi: 10.1007/BF02235139
-
[5]
ROSS S D, BLESSING J E. Alcohols as H-donor media in coal conversion. 2. Base-promoted H-donation to coal by methyl alcohol[J]. Fuel, 1979, 58(6): 438-442. doi: 10.1016/0016-2361(79)90085-1
-
[6]
LEI Z, LIU M, SHUI H. Study on the liquefaction of Shengli lignite with NaOH/methanol[J]. Fuel Process Technol, 2010, 91(7): 783-788. doi: 10.1016/j.fuproc.2010.02.014
-
[7]
KUZNETSOV P N, SHARYPOV V I, BEREGOVTSOVA N G. Properties of Kansk-Atchinsk lignite during liquefaction in lower alcohols[J]. Fuel, 1990, 69(7): 911-916. doi: 10.1016/0016-2361(90)90241-H
-
[8]
MONDRAGON F, ITOH H, OUCHI K. Solubility increase of coal by alkylation with various alcohols[J]. Fuel, 1982, 61(11): 1131-1134. doi: 10.1016/0016-2361(82)90198-3
-
[9]
LU H Y, WEI X Y, YU R. Sequential thermal dissolution of huolinguole lignite in methanol and ethanol[J]. Energy Fuels, 2011, 25(6): 2741-2745. doi: 10.1021/ef101734f
-
[10]
ZIEGLER T, AUTSCHBACH J. Theoretical methods of potential use for studies of inorganic reaction mechanisms[J]. Chem Rev, 2005, 105(6): 2695-2722. doi: 10.1021/cr0307188
-
[11]
GRIMME S. Calculation of frequency dependent optical rotation using density functional response theory[J]. Chem Phys Lett, 2001, 339(5/6): 380-388.
-
[12]
IE Y, HIROSE T, NAKAMURA H. Nature of electron transport by pyridine-based tripodal anchors: Potential for robust and conductive single-molecule junctions with gold electrodes[J]. J Am Chem Soc, 2011, 133(9): 3014-3022. doi: 10.1021/ja109577f
-
[13]
GORELSKY S I, LEVER A B P. Electronic structure and spectra of ruthenium diimine complexes by density functional theory and INDO/S. Comparison of the two methods[J]. J Organomet Chem, 2011, 635(1/2): 187-196.
-
[14]
GEERLINGS P, AYERS P W, TORO-LABBÉ A. The woodward-offmann rules reinterpreted by conceptual density functional theory[J]. Acc Chem Res, 2012, 45(5): 683-695. doi: 10.1021/ar200192t
-
[15]
JIANG Z, PAN Q, LI M. Density functional theory study on direct catalytic decomposition of ammonia on Pd (111) surface[J]. Appl Surf Sci, 2013, 292: 494-499.
-
[16]
HUANG J, LIU C, REN L, TONG H, LI W, WU D. Studies on pyrolysis mechanism of syringol as lignin model compound by quantum chemistry[J]. J Fuel Chem Technol, 2013, 41(6): 657-666. doi: 10.1016/S1872-5813(13)60031-6
-
[17]
ZHANG X, GAO H, XU H. A density functional theory study of the hydrolysis mechanism of phosphodiester catalyzed by a mononuclear Zn (Ⅱ) complex[J]. J Mol Catal A: Chem, 2012, 368-369: 53-60.
-
[18]
SHIM J G., KIM J H, JHON Y H. DFT calculations on the role of base in the reaction between CO2 and monoethanolamine[J]. Ind Eng Chem Res, 2009, 48(4): 2172-2178. doi: 10.1021/ie800684a
-
[19]
AZIZPOUR H, SOTUDEH-GHAREBAGH R, MOSTOUFI N. Characterization of regime transition in fluidized beds at high velocities by analysis of vibration signals[J]. Ind Eng Chem Res, 2012, 51(7): 2855-2863. doi: 10.1021/ie200863y
-
[20]
WANG W J, CAO Y Y. Theoretical study of ethanol partial oxidation for syngas production under cold plasma conditions[J]. J Energy Inst, 2014, 87(2): 89-95. doi: 10.1016/j.joei.2014.03.026
-
[21]
KYUNGBOOK L, HOONYOUNG J, SEUNGPIL J, JONGGEUN C. Improvement of ensemble smoother with clustered covariance for channelized reservoirs[J]. Energy Explor Exploit, 2013, 31(5): 713-726. doi: 10.1260/0144-5987.31.5.713
-
[22]
NAJAFI M, MOOD K H, ZAHEDI M. DFT/B3LYP study of the substituent effect on the reaction enthalpies of the individual steps of single electron transfer-proton transfer and sequential proton loss electron transfer mechanisms of chroman derivatives antioxidant action[J]. Comput Theor Chem, 2011, 969(1/3): 1-12.
-
[23]
WANG H, SHI X, CHE D. Thermodynamic optimization of the operating parameters for a combined power cycle utilizing low-temperature waste heat and LNG cold energy[J]. Appl Therm Eng, 2013, 59(1/2): 490-497.
-
[24]
ZHOU J, ZONG Z M, CHEN B. The Enrichment and identification of methyl alkanones from thermally soluble shengli lignite[J]. Energy Source Part A, 2013, 35(23): 2218-2224. doi: 10.1080/15567036.2011.652759
-
[25]
PICCOLO A, SPACCINI R, NIEDER R. Sequestration of a biologically labile organic carbon in soils by humified organic matter[J]. Clim Change, 2004, 67(2): 329-343.
-
[26]
ADLER E. Lignin chemistry-past, present and future[J]. Wood Sci Technol, 1977, 11(3): 169-218. doi: 10.1007/BF00365615
-
[27]
YU L C, WEI X Y, WANG Y H. Catalytic hydroconversion of extraction residue from Shengli lignite over Fe-S/ZSM-5[J]. Fuel Process Technol, 2014, 126: 131-137. doi: 10.1016/j.fuproc.2014.04.032
-
[28]
JUG K. A new definition of atomic charges in molecules[J]. Theor Chem Acta, 1973, 31(1): 63-73. doi: 10.1007/BF00527439
-
[29]
XU X, WANG Y, CHEN Z, BAI L, ZHANG K, YANG S, ZHANG S. Influence of cooling treatments on char microstructure and reactivity of Shengli brown coal[J]. J Fuel Chem Technol, 2015, 43(1): 1-8. doi: 10.1016/S1872-5813(15)60005-6
-
[30]
LUNDBERG M, BOROWSKI T. Oxoferryl species in mononuclear non-heme iron enzymes: Biosynthesis, properties and reactivity from a theoretical perspective[J]. Coord Chem Rev, 2012, 257(1): 277-289.
-
[31]
GECE G, BILGIC S. Molecular-level understanding of the inhibition efficiency of some inhibitors of zinc corrosion by quantum chemical approach[J]. Ind Eng Chem Res, 2012, 51(43): 14115-14120. doi: 10.1021/ie302324b
-
[1]
-

Figure 3 Thermodynamic properties of components
(a):BOB; (b):isopropanol; (c):BO; (d):phenol
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
