School of Pharmacy, China Pharmaceutical University, Nanjing 211198, China
b.
Department of Geriatrics, The Second Affiliated Hospital, Nanjing Medical University, Nanjing 210011, China
c.
Key Laboratory for Aging & Disease, Nanjing Medical University, Nanjing 210011, China
d.
Jiangsu Aosaikang Pharmaceutical Co., Ltd., Nanjing 211112, China
e.
Department of Respiratory Disease, Baoshan Branch, Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 200120, China
f.
Department of Geriatric Cardiology, Jiangsu Provincial Key Laboratory of Geriatrics, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China
g.
Department of Geriatrics, The Affiliated Sir Run Run Hospital of Nanjing Medical University, Nanjing 211166, China
* Corresponding authors. ** Corresponding author at: Department of Geriatrics
weihe@cpu.edu.cn (W. He). 1 These authors contributed equally to this work.
Received Date:
27 December 2021 Accepted Date:
28 April 2022 Revised Date:
27 April 2022 Available Online:
15 January 2023
Abstract:
Plaque plays a central role in atherosclerosis (AS) progression, whereas inflammation and destruction of the plaque microenvironment contribute to plaque advancement. As a result, a therapy regime, which combines anti-inflammation and inhibition-degradation of plaque matrix, appears to be a promising strategy to combat AS. Herein, we report a pH-sensitive liposome co-loading with the anti-inflammatory agent (oridonin, ORD) and plaque-collagen protector (marimastat) for anti-AS therapy. ORD was first conjugated with hyaluronic acid (HA) to target the inflammation contributor, pro-inflammatory macrophages. Then, the conjugate assembled onto the MATT-loaded liposomes. The co-loaded system (~150 nm) significantly improved pharmacokinetics over the liposomes without anchoring the conjugate and accumulated effectively in the plaque. The preparation administration allowed efficient anti-AS activities in high-fat diet (HFD)-Apoe-/- mice by decreasing the pro-inflammatory cytokine expression in the serum, lessening the lesion area, alleviating the plaque collagen degradation, promoting macrophage polarization from phenotypic M1 to M2, reducing T helper (Th) 17 cells (Th17)/T regulatory cells (Tregs) and Th1/Th2 ratio, etc. Furthermore, the serum determination in AS patients demonstrated high expression of the inflammatory cytokines, indicating our finding may offer a potential guideline for clinical practice.
Atherosclerosis (AS) is a chronic inflammatory disease characterized by plaque buildup at the arterial subendothelial space primarily owing to the deposition of activated cells such as macrophages and endothelial cells, lipid and extracellular matrix-like collagen, and glycosaminoglycan [1-4]. Clinically, the rupture of the plaque frequently leads to severe cardiovascular events, such as acute myocardial infarction (AMI), stroke, and peripheral arterial disease [5], accounting for 30% of cardiac death worldwide [6]. Statin therapy for lipid-lowering is the most commonly used for AS treatment [7, 8]. However, this regimen has a modest effect on plaque progression [9], and inversely, may increase the risk of coronary heart disease [10].
Constant inflammation reaction is a vital determinant of plaque development [11]. Whereas macrophages, the most abundant leukocytes in the plaque, significantly contribute to inflammation response via the production of pro-inflammatory cytokines [12]. Unfortunately, single anti-inflammation therapy remains not reduce AS patient mortality [13]. Furthermore, the degradation of extracellular plaque-matrix, which disrupts the plaque microenvironment, is another essential factor in promoting plaque advancement [14, 15]. Collagen constitutes a central portion of the plaque matrix and forms a structural framework under the fibrous cap [16]. Via providing strength and integrity for the fibrous cap, collagen can protect the plaque from breakup [17]. Accordingly, a combined regimen to inhibit decomposition of plaque collagen and suppress inflammation via targeting plaque macrophages appears to be a promising approach for plaque stabilization.
Liposomes have been widely used as drug carriers to improve drug delivery due to their various benefits. These include increasing stability through encapsulation, increasing drug efficacy and therapeutic index, improving pharmacokinetic effects (i.e., decrease elimination and increase circulation lifetime), and reducing the toxicity of encapsulated agents [18, 19]. This study developed a pH-sensitive liposomal system to co-deliver an anti-inflammatory drug (oridonin, ORD) and a plaque-collagen protector (marimastat, MATT) to the plaque. Briefly, ORD is an anti-inflammatory compound with poor water solubility [20]. To improve its solubility and target macrophages, ORD was modified with hyaluronic acid (HA). The HA in the conjugate (HA-ORD) could target cell CD44 receptors [21]. MATT, a water-soluble compound, is an effective protector of collagen [22], and MATT could inhibit plaque-collagen degradation by matrix metalloproteinases (MMPs) [23]. The pH-sensitive co-delivery liposomes, termed co-loaded pHSLs, were prepared by encapsulating the collagen protector into the aqueous cores and anchoring the conjugate onto the liposomes (Fig. 1A). The plaque has an over-metabolism and the resultant acidic microenvironment [24]. We proposed that the co-loaded pHSLs could release the two therapeutics in response to the acidic microenvironment after accumulating in the plaque, thereby achieving efficient anti-AS therapy. To obtain proof of concept, we conducted various experiments in vitro and in vivo. Apoe-/- mice fed with a high-fat diet (HFD) were utilized as a model in the anti-AS study in vivo.
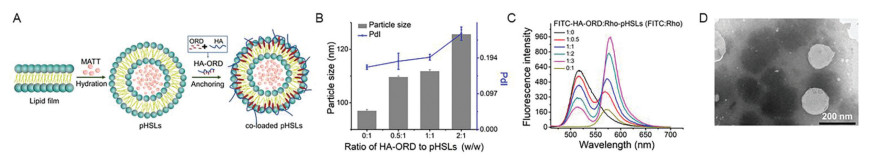
Figure 1
Figure 1.
Preparation and characterization. (A) Iillustration of the co-loaded pHSLs preparation. (B) Influence of HA-ORD/lipid mass ratio on the pHSLs diameter. (C) FRET between FITC-HA-ORD/Rho-pHSLs with different FITC and Rho mass ratios (excitation wavelength: 430 nm). The HA-ORD was conjugated with FITC, and Rho was loaded in the pHSLs cores. (D) TEM examination.
The pH-sensitive co-delivery liposomes (co-loaded pHSLs) were prepared by assembling the conjugate HA-ORD onto the MATT-loaded pH-sensitive liposomes (MATT-pHSLs) (Fig. 1A). First, the targeted conjugate hyaluronic acid-oridonin (HA-ORD) was synthesized via condensation reaction. The synthesis of HA-ORD was characterized by 1H and 13C NMR spectra. Consequently, approximately 70% of fed ORD was linked to HA. Five ORD molecules were involved in one conjugate. MATT-pHSLs were prepared by the film dispersion method and had a diameter of 100–120 nm. Then, the co-loaded pHSLs were prepared by anchoring the hydrophobic section of conjugate onto the liposomes via hydrophobic interactions under ultrasonic conditions. As shown in Fig. 1B, the increased proportion of HA-ORD/lipid (w/w) increased the particle size of co-loaded pHSLs from 90 nm to 130 nm. To verify the conjugate loading onto the liposomes, we performed the FRET test by labeling the conjugate with the fluorescence donor (FITC) and encapsulating the fluorescence acceptor (Rho) in the cores of liposomes. As a result of an increase in the mass ratio of FITC/Rho, the maximum intensity of the fluorescence acceptor (Rho) at 575 nm rises gradually, whereas the maximal fluorescence intensity of the donor (FITC) at 515 nm declines (Fig. 1C). The results demonstrated the happening of the FRET effect. FRET is always utilized to explore the interaction between two materials by tagging fluorescent donors and acceptors to them [25-27]. These results implied the co-assembly of the conjugate and the liposomes. TEM examination displayed the co-loaded pHSLs have a spherical morphology with a 150 nm diameter (Fig. 1D). The incubation in 10% serum for 6 h imposed little change on the particle size of MATT-pHSLs and co-loaded pHSLs and, therefore, indicated potential stability in the blood after intravenous injection (Fig. S1 in Supporting information). In addition, little FRET effect between the 6 h incubation liposomes and 0 h incubation samples, implying the two payloads were loaded in the liposomes after serum incubation (Fig. S2 in Supporting information). HPLC assay demonstrated that total drug-loading in the co-loaded pHSLs was 8.27% ± 0.19%, wherein the MATT loading was 2.35% ± 0.20% with an encapsulation efficiency of 72.78% ± 6.27%, and while the ORD loading was 6.06%, respectively. After 24 h incubation in 10% serum, the remaining drugs (%) were 98.68% MATT and 99.85% ORD in the co-loaded pHSLs compared with the initial drug loading, demonstrating little drug leakage. The release study depicted that the release of MATT and HA-ORD conjugate from the liposomes at pH 6.0 in first 1 h period was significantly greater than that at pH 7.4 (Fig. S3 in Supporting information, P < 0.05), thereby verified the pH-sensitive characteristics of the liposomes.
To study anti-AS in vitro, we performed various experiments, including M1-like macrophage-targeting, uptake of ox-LDL in macrophages and inflammation inhibition. The accumulation of pro-inflammatory macrophages (M1-like phenotype) promotes the AS pathological process [28]. We expected that delivering the targeted anti-inflammatory agent, HA-ORD, to the M1-like macrophages could retard plaque progression through regression of ox-LDL uptake and inflammation activity. First, the targeted uptake of HA-ORD by M1-like macrophages induced by LPS was assayed. The CD44 expression on the M1-like macrophages was verified by confocal imaging after staining with the FITC-CD44 antibody (Fig. S4 in Supporting information). To study the conjugate target ability to the CD44 receptor, we saturated the receptor on the M1-like macrophages by incubation with a high HA-concentration. As shown in Figs. S5A and B (Supporting information), the Rho-HA-ORD uptake in the HA-saturated cells declined significantly (P < 0.01). Furthermore, the uptake in the M1-like macrophages was significantly higher than that in nonpolarized RAW264.7 (Figs. S5C and D in Supporting information). In addition, the M1-like macrophages uptake was time-related and reached maximal internalization at 2 h post-incubation (Fig. S6 in Supporting information). Overall, the data implied that the conjugate (HA-ORD) could target M1-like macrophages.
The excessive influx of ox-LDL facilitates the formation of the foam cells and then induces the development of plaques [29]. Accordingly, the inhibition of ox-LDL uptake in macrophages functionally impedes AS progress and promotes plaque stability [30, 31]. As depicted in Figs. S7A and B (Supporting information), the M1-macrophages (LPS-induced RAW264.7) possess improved ability to take up ox-LDL over the nonpolarized RAW264.7 without LPS-treatment (Figs. S7A and B, Fig. S8 in Supporting information). The treatment with co-loaded pHSLs restrained the ox-LDL influx with higher efficacy than the incubation with other formulations (Fig. S7B, P < 0.001). Furthermore, the anti-inflammatory study indicated that HA-ORD-containing formulations, HA-ORD and co-loaded pHSLs, reduced the production of inflammatory cytokines, IL-6 and TNF-α (Figs. S7C and D in Supporting information), while co-loaded pHSLs allowed enhanced anti-inflammation effect. These results demonstrated that co-loaded pHSLs could alleviate AS in vitro.
Next, we investigated anti-AS efficacy in vivo in the Apoe-/- mice with HFD feed via assaying plaque volume and blood lipid level. After 4 weeks of HFD feeding, the Apoe-/- mice were intravenously administrated with different formulations every five days for 8 weeks (Fig. S9A in Supporting information). We first stained descending aorta with oil-red-O (ORO) to assay the lesion area after treatment. As displayed in Figs. S9B and C (Supporting information), combination treatment with the physical mixture or the co-loaded pHSLs reduced the lesion area in the descending aorta compared with the single treatment of free MATT or HA-ORD (P < 0.01). Significantly, the dosing of co-loaded pHSLs at low and high doses rendered 0.5 (P < 0.05) and 1-fold decrease (P < 0.01) in the lesion area over the single administration. Next, we determined the plaque area by staining the aortic sinus cryosection with H&E and ORO (Figs. S9D and E in Supporting information). As showed in H&E and ORO staining, the lesions were inhibited after therapy. The plaque area in the group treated with the co-loaded pHSLs was reduced compared with the groups dosed with the PBS, free HA-ORD, or physical mixture of MATT and HA-ORD (Fig. S9D). The semi-quantitative analysis indicated that the plaque area in groups dosed with PBS, free MATT, free HA-ORD, physical mixture, co-loaded pHSLs at low and high doses was 32.34% ± 1.96%, 31.77% ± 0.63%, 26.10% ± 3.30%, 25.20% ± 2.40%, 17.33% ± 4.72% and 12.34% ± 2.11%, respectively (Fig. S9E). The treatment with the co-loaded pHSLs enabled a 1–1.5-fold plaque-area decrease over the PBS treatment. In addition, the mouse weight usually increased during the treatment duration. As a result, no apparent pathologic change after treatment was observed by H&E staining, indicating the safety of these formulations (Figs. S9F and S10 in Supporting information). Overall, the results demonstrated that the therapy with the co-loaded pHSLs could impede plaque advancement effectively.
Blood lipid is a significant factor contributing to AS progression, including low-density lipoproteins (LDL), cholesterol, high-density lipoproteins (HDL) cholesterol, and other components [32]. As reported, the HDL protected the HFD diet animals from AS and, inversely, the LDL is a kind of "bad" cholesterol that its accumulation accelerates the development of AS [33, 34]. First, we determined the blood lipid levels after dosing. The treatment with the co-loaded pHSLs lowered the serum LDL and total cholesterol (TC) compared with PBS-treatment (P < 0.05), accompanied with a modest effect on triglyceride (TG) and HDL (Figs. S11A–D in Supporting information). Next, we calculated the plasma TG/HDL ratio and atherogenic index (common logarithm of TC/HDL ratio). The treatment with co-loaded pHSLs reduced the ratio of TG/HDL and the atherogenic index compared with PBS treatment (Figs. S11E and F in Supporting information, P < 0.05). The increase in the TG/HDL ratio and atherogenic index indicates AS exacerbation and a growing risk of cardiovascular events [35]. As a result, the co-loaded pHSLs administration is promising to attenuate AS by lowering the lipid levels.
To investigate the contributors to the effective anti-AS efficacy, we next studied the pharmacokinetics of co-loaded pHSLs and explored their targetability to the active axis of plaque-microenvironment components (macrophages and collagen)-inflammatory factors.
Blood circulation is an essential factor that governs the accumulation of drug carriers in the target site of interest [36, 37]. And the fluorescence dye was widely used to quantify the pharmacokinetics of carriers, especially the environment-responsive dye, such as aggregation-caused quenching (ACQ) probe [38, 39]. Furthermore, previous reports demonstrated that the carrier circulation performance obtained by determining the drug was generally similar to fluorescence-dye determination [40]. As depicted in Fig. 2A, the DiR labeled co-loaded pHSLs displayed significantly higher plasma concentrations at each time point post-injection than DiR-pHSLs without anchoring of HA-ORD. Next, the pharmacokinetic parameters were calculated based plasma concentration (Table S1 in Supporting information). The co-loaded pHSLs demonstrated prolonged t1/2 of blood circulation and mean residence time (P < 0.05), increased AUC (P < 0.05), and lowered clearance rate (CL, P < 0.05) in contrast to pHSLs. Therefore, the data indicated that the conjugate (HA-ORD) anchoring enabled prolonged blood circulation time and improved pharmacokinetics. The improved performance in vivo of co-loaded pHSLs may be because the surface HA could offer an extra energy barrier for nanoparticles by presenting bound water molecules and compromising the absorption of opsonin [41, 42].
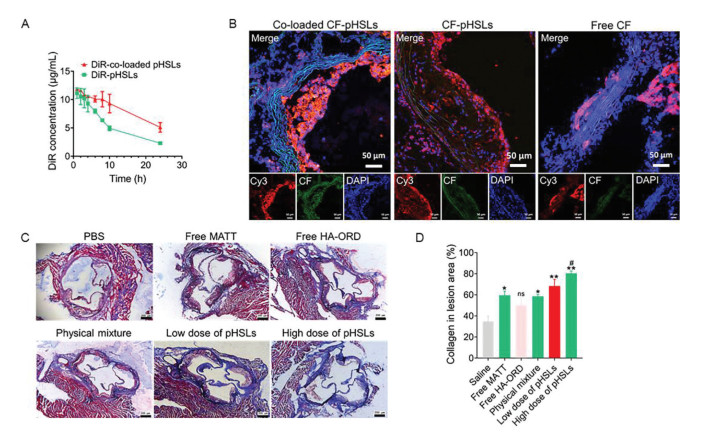
Figure 2
Figure 2.
Pharmacokinetics, plaque targeting and collagen in the lesion area. (A) Pharmacokinetics of pHSLs and co-loaded pHSLs. DiR labeled formulations were intravenously injected at a DiR dose of 5 mg/kg, according to the body weight. The fluorescence intensity was detected by a microplate reader at 748 nm (excitation) and 780 nm (emission). n = 5. (B) Targeting the plaque in Apoe-/- HFD mice. The tricuspid ring of the mouse heart was isolated for sectioning after 2 h injection with CF-labeled formulations and the control (free CF) via the tail vein at a CF dose of 2.5 mg/kg, according to the body weight. The plaque was marked by labeling the CD68 on macrophages with Cy3 (red, 5 µg/mL). Scale bar: 50 µm. (C) Masson staining of the descending aortas after administration. The collagen fibers were stained to blue and muscle fibers to red. The plaque was stained to red. The dosage of ORD and MATT in the pHSLs was 5/5 mg/kg and 5/10 mg/kg for low dose and high dose, respectively. (D) Semi-quantitative of collagen in the plaque calculated by ImageJ. pHSLs herein were the co-loaded pHSLs. Scale bar: 200 µm. n = 3, P < 0.05, **P < 0.01, compared with saline; #P < 0.05, compared with free MATT; ns, not significant.
Then, we first studied the plaque-targeted ability of CF-labeled co-loaded pHSLs in the Apoe-/- HFD model. As well known, numerous M1-like macrophages inhabit inside the plaque [43]. Accordingly, the plaque targeting was characterized by co-localizing the CF-labeled pHSLs with the M1-like macrophages marked by Cy3 for CD68. As shown in Fig. 2B, profound yellow fluorescent spots were exhibited in the group dosed with the CF-labeled pHSLs or co-loaded pHSLs; in contrast, slight yellow fluorescence was observed in the group treated with free dye. Importantly, co-loaded pHSLs have more profound co-localization compared with CF-labeled pHSLs. The results demonstrated that the co-loaded pHSLs with HA-ORD anchoring effectively accumulated at the plaque and located inside the lesion area.
Collagen, a significant component of the plaque extracellular matrix, is critical to stabilizing the plaque and preventing rupture [44]. The higher the collagen content, the more stable the plaque [45]. Herein, the collagen was identified by Masson staining. As shown in the Masson staining (Fig. 2C), the positive collagen stained in blue was demonstrated in the groups treated with MATT-loading preparations, especially with co-loaded pHSLs at low or high doses. Quantified assay indicated that the treatment with co-loaded pHSLs allowed a 1–1.5-fold increase of collagen expression compared to PBS treatment (Fig. 2D, P < 0.01). Furthermore, co-loaded pHSLs (high dose) demonstrated elevated collagen expression compared to the physical mixture (P < 0.05). These results verified that the co-loaded pHSLs could alleviate the plaque-collagen degradation and, therefore, is promising to protect the plaque from disruption.
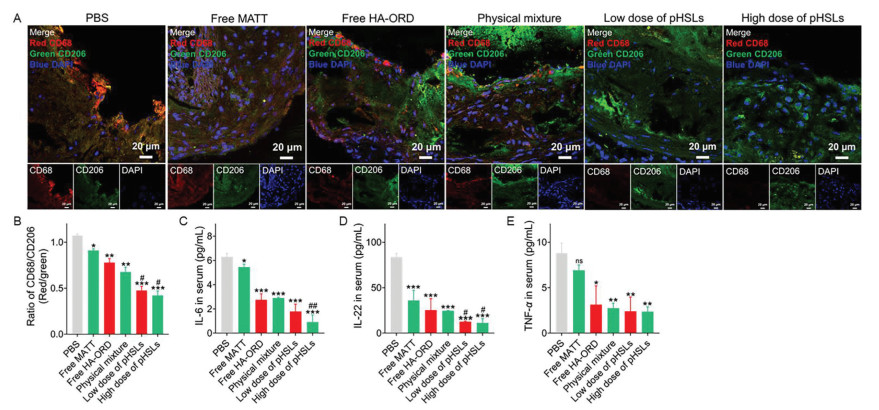
To study macrophage polarization switch, we first assayed the polarization pattern of macrophages in the plaque in Apoe-/- HFD model after treatment. As depicted in the result of confocal imaging (Fig. 3A), a large amount of red fluorescent spots (marking CD68+) and slight green fluorescence (marking CD206+) appeared in the PBS group. Conversely, we observed fewer red fluorescent spots and considerable green fluorescence in the groups administered with HA-ORD loading preparations. Notably, co-loaded pHSLs demonstrated elevated green fluorescence intensity compared to the physical mixture (PM) and other preparations. Additional quantified assay indicated that the ratio of CD68+/CD206+ macrophages from the groups administered with co-loaded pHSLs at the low or high dose displayed approximately 80% and 40% reduction compared with the PBS- and PM-treated groups, respectively (Fig. 3B, P < 0.001 and P < 0.05). To confirm the phenotype switch of macrophages, we also assayed the M1/M2-like subset in the liver after treatment. The administration of co-loaded pHSLs did not affect the total number of liver macrophages as compared with PBS (Figs. S12A and B in Supporting information), whereas the administration resulted in the expansion of CD206+ macrophages and reduction of CD68+ phenotype (Figs. S12C and D in Supporting information, P < 0.05).
Figure 3
Figure 3.
Targeting macrophages and ameliorating inflammation. (A) Analysis of macrophages by immunofluorescence staining and (B) semi-quantitative of the M1/M2 ratio by ImageJ. M1 macrophages (CD68) were stained to red with Cy3-labeled second antibody, and M2 macrophages (CD206) were stained to green with FITC-labeled second antibody. Scale bar: 20 µm. (C–E) The inflammatory cytokines expression in the serum. IL-6 and TNF-α were measured using the Bio-Plex® multiplex array systems. IL-22 was determined with ELISA kits. The dosage for ORD and MATT was 5 mg/kg. The low dose and high dose indicate the different dosages of co-loaded pHSLs, 5/5 mg/kg and 5/10 mg/kg for ORD and MATT. n = 3, P < 0.05, **P < 0.01, ***P < 0.001, compared with saline; #P < 0.05, ##P < 0.01, compared with free HA-ORD, free MATT or physical mixture; ns, not significant.
In addition, we detected the immune cells, T regulatory cells (Tregs) and T-lymphocyte helper (Th) cells, in the serum, owing to that Tregs could inhibit inflammation and AS progression, while the increased ratio of Th17/Tregs and Th1/Th2 always exacerbate inflammation and atherogenesis [46-48]. As displayed in Figs. S13A–D (Supporting information), dosing the co-loaded pHSLs at low or high dose demonstrated limited influence on the Treg percent and, while reducing the ratio of Th17/Tregs (P < 0.05) and Th1/Th2 (P < 0.01) compared with PBS administration. Overall, the data revealed the treatment with co-loaded pHSLs regressed macrophage polarization switch towards pro-inflammatory phenotype in the plaque and livers and decreased the ratio of Th1/Tregs and Th1/Th2, which could benefit the plaque stabilization [49].
Finally, the inflammatory serum cytokines were measured after treatment. The pro-inflammatory factors, IL-6, IL-22, and TNF-α, were significantly downregulated after treatment with HA-ORD loading preparations, including free HA-ORD, PM, and co-loaded pHSLs, compared with PBS treatment (Figs. 3C–E, P < 0.001). Especially, the dosing of co-loaded pHSLs at a high dose reduced the levels of IL-6 and IL-22 by 3- and 1.5-fold, respectively, over PM treatment (Figs. 3C–E). The data implied that the treatment with co-loaded pHSLs could lessen the inflammation.
To confirm the present results that might provide potential guidelines for the clinical treatment, we further tested the serum cytokines in patients with acute myocardial infarction (AMI) caused by the rupture of AS plaque [50] and healthy volunteers without cardiovascular disease. The population baseline table is shown in Table S2 (Supporting information). As displayed in Fig. S14 (Supporting information), elevated inflammatory cytokines, IL-6, IL-22, and TNF-α, were demonstrated in the patients compared with the healthy volunteers (P < 0.001). The data indicated that our results could be translated into the clinic practices of AS therapy.
Taken together, we proved that the co-loaded pHSLs had extended blood circulation time, effectively accumulated in the plaque, facilitated macrophage polarization into M2-like phenotype, and rendered inhibition of both inflammation and collagen degradation (Fig. S15 in Supporting information). Furthermore, their administration to HFD-Apoe-/- mice reduced the lesion area, elevated the percent of Tregs, decreased the Th1/Th2 ratio. Targeted co-delivery of inflammation inhibitor and collagen protector to the plaque represents a promising approach for anti-AS therapy.
The anchoring strategy developed here acts as a promising approach to load a polymer-based conjugate/prodrug in liposomes to improve delivery. The conjugate HA-ORD with a molecular weight of over 10,000 Da is challenging to be loaded in liposome core or lipid bilayer. Rather than loading the conjugate at the two locations, we loaded it on liposomes by the anchoring strategy that the hydrophobic section in conjugate inserted into the liposome's lipid membrane. Furthermore, the anchoring improved the pharmacokinetic performance of the pHSLs. In addition, the anchoring facilitated plaque accumulation of the co-loaded pHSLs because HA on the co-loaded pHSLs could target the CD44 receptors that are overexpressed on the cells inhabiting the plaques. The CD44 receptors in the plaque have > 10-fold increase over that in healthy vascular tissue [51]. Nonetheless, the plaque targeting-efficacy of drug delivery systems is correlated with plaque advancement [52]. Additional study is needed to clarify the targetability of the co-loaded pHSLs to the early and advanced plaque.
The features, including thin fibrous cap, inflammation, accumulation of inflammatory cells, and raised blood lipid levels are closely linked to the rupture of the plaque [53]. Significantly, the loss of fibrillar collagen is a direct cause to make the fibrous cap become thinning [54]. In this study, we found that the targeting co-delivery of the collagen protector MATT and anti-inflammatory HA-ORD conjugate using pHSLs to the plaque relieved the degradation of plaque collagen, reduced the accumulation of M1-like macrophages, alleviated inflammation, and declined the blood level. These indicators confirmed that the co-loaded pHSLs could stabilize the rupture-prone plaque in HFD-Apoe-/- mice. Furthermore, we found that the plaque was regressed, as evidenced by dosing the co-loaded pHSLs to HFD-Apoe-/- mice reduced the plaque area by 1–1.5-fold compared to the PBS treatment. The plaque regression may be ascribed to the lowering of blood lipids, LDL and TC, and reduced residence of pro-inflammatory macrophages after administration of co-loaded pHSLs [55].
IL-6 plays a leading role in the inflammatory progressions underlying cardiovascular diseases [28, 56], and meanwhile, TNF-α facilitates the development of plaque at all stages [12, 57]. Herein, we demonstrated that treatment with the co-loaded pHSLs significantly reduced the IL-6, IL-22, and TNF-α in HFD-Apoe-/- mice (Figs. 3C–E). The data is consistent with the serum determination from the AS patients, in which the two indicators markedly upregulated, as displayed in Fig. S14. As a result, our data may inspire the clinic treatment of AS, which is combinatorial use of anti-inflammatory drugs and collagen protectors to improve AS therapy.
In summary, the co-loaded pHSLs allowed effective anti-AS therapy by targeted co-delivery of the anti-inflammatory (HA-ORD) and collagen protector (MATT) to the plaque. Moreover, the targeted co-delivery suppressed plaque regression. Besides promoting macrophage polarization into M2-like phenotype and alleviating inflammation and plaque-collagen degradation, we confirmed that the co-delivery could lower the blood lipid level and modulate the immune-cell ratio of Th17/Tregs and Th1/Th2 as well. We believe the current liposomal strategy is promising to target the AS plaque. Targeted co-delivery of anti-inflammatory drugs and collagen protectors to the plaque represents an efficient regimen to treat AS. The finding may provide a potential guideline for the clinic practices of AS treatment.
MATT utilized in the study was a broad-spectrum MMP inhibitor and was not approved for clinical use. However, Periostat, an MMP inhibitor, has been permitted clinically [58]. Accordingly, the combinatorial use of an anti-inflammatory drug and Periostat has the potential as an AS therapy regimen.
Declaration of competing interest
The authors have no conflicts of interest to declare.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Nos. 81872823, 82073782, 81970374), the Shanghai Science and Technology Committee (No. 19430741500). We are thankful for the guidance and help provided by Lab Center, the Second Affiliated Hospital of Nanjing Medical University.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.04.081.
[1]
N.V.K. Pothineni, S. Subramany, K. Kuriakose, et al., Eur. Heart J.38 (2017) 3195-3201. doi: 10.1093/eurheartj/ehx362
Figure 1
Preparation and characterization. (A) Iillustration of the co-loaded pHSLs preparation. (B) Influence of HA-ORD/lipid mass ratio on the pHSLs diameter. (C) FRET between FITC-HA-ORD/Rho-pHSLs with different FITC and Rho mass ratios (excitation wavelength: 430 nm). The HA-ORD was conjugated with FITC, and Rho was loaded in the pHSLs cores. (D) TEM examination.
Figure 2
Pharmacokinetics, plaque targeting and collagen in the lesion area. (A) Pharmacokinetics of pHSLs and co-loaded pHSLs. DiR labeled formulations were intravenously injected at a DiR dose of 5 mg/kg, according to the body weight. The fluorescence intensity was detected by a microplate reader at 748 nm (excitation) and 780 nm (emission). n = 5. (B) Targeting the plaque in Apoe-/- HFD mice. The tricuspid ring of the mouse heart was isolated for sectioning after 2 h injection with CF-labeled formulations and the control (free CF) via the tail vein at a CF dose of 2.5 mg/kg, according to the body weight. The plaque was marked by labeling the CD68 on macrophages with Cy3 (red, 5 µg/mL). Scale bar: 50 µm. (C) Masson staining of the descending aortas after administration. The collagen fibers were stained to blue and muscle fibers to red. The plaque was stained to red. The dosage of ORD and MATT in the pHSLs was 5/5 mg/kg and 5/10 mg/kg for low dose and high dose, respectively. (D) Semi-quantitative of collagen in the plaque calculated by ImageJ. pHSLs herein were the co-loaded pHSLs. Scale bar: 200 µm. n = 3, P < 0.05, **P < 0.01, compared with saline; #P < 0.05, compared with free MATT; ns, not significant.
Figure 3
Targeting macrophages and ameliorating inflammation. (A) Analysis of macrophages by immunofluorescence staining and (B) semi-quantitative of the M1/M2 ratio by ImageJ. M1 macrophages (CD68) were stained to red with Cy3-labeled second antibody, and M2 macrophages (CD206) were stained to green with FITC-labeled second antibody. Scale bar: 20 µm. (C–E) The inflammatory cytokines expression in the serum. IL-6 and TNF-α were measured using the Bio-Plex® multiplex array systems. IL-22 was determined with ELISA kits. The dosage for ORD and MATT was 5 mg/kg. The low dose and high dose indicate the different dosages of co-loaded pHSLs, 5/5 mg/kg and 5/10 mg/kg for ORD and MATT. n = 3, P < 0.05, **P < 0.01, ***P < 0.001, compared with saline; #P < 0.05, ##P < 0.01, compared with free HA-ORD, free MATT or physical mixture; ns, not significant.

 DownLoad:
DownLoad:

 下载:
下载:




